Nowe publikacje
Nowe odkrycia przyczyniają się do lepszego zrozumienia przyczyn zespołu Retta
Ostatnia recenzja: 02.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.

Zespół Retta to rzadkie zaburzenie neurorozwojowe, na które obecnie nie ma lekarstwa ani dobrego leczenia. Powoduje poważne objawy fizyczne i poznawcze, z których wiele pokrywa się z zaburzeniami ze spektrum autyzmu.
Zespół Retta jest spowodowany mutacjami w genie MECP2, który jest silnie ekspresjonowany w mózgu i wydaje się odgrywać ważną rolę w utrzymaniu zdrowia neuronów. Gen znajduje się na chromosomie X, a zespół ten dotyka głównie dziewczynki. Aby opracować metody leczenia zespołu Retta, naukowcy chcą lepiej zrozumieć MECP2 i jego funkcje w mózgu.
Naukowcy, w tym współzałożyciel Whitehead Institute Rudolf Jaenisch, badają MECP2 od dziesięcioleci, jednak wiele podstawowych faktów na temat genu pozostało nieznanych. Białko kodowane przez gen, MECP2, bierze udział w regulacji genów; wiąże się z DNA i wpływa na poziom ekspresji różnych innych genów lub ilość białka, które produkują.
Naukowcy nie dysponowali jednak kompletną listą genów, na które wpływa MECP2, i nie było konsensusu co do tego, w jaki sposób MECP2 wpływa na te geny.
Wczesne badania MECP2 sugerowały, że jest represorem, zmniejszającym ekspresję swoich docelowych genów, ale badania Jaenischa i innych wykazały wcześniej, że MECP2 działa również jako aktywator, zwiększając ekspresję swoich celów — i że może być aktywatorem na pierwszym miejscu. Nieznany był również mechanizm działania MECP2 ani to, co dokładnie białko robi, aby powodować zmiany w ekspresji genów.
Ograniczenia technologiczne uniemożliwiły badaczom uzyskanie jasności w tych kwestiach. Ale Yanish, jego pracownik naukowy Yi Liu i były członek laboratorium Yanisha Anthony Flamier, obecnie adiunkt w ośrodku badawczym CHU Sainte-Justine na Université de Montréal, wykorzystali najnowocześniejsze techniki, aby odpowiedzieć na pozostałe pytania dotyczące MECP2 i uzyskać nowe spostrzeżenia na temat jego roli w zdrowiu i chorobach mózgu.
Wyniki ich badań opublikowano w czasopiśmie Neuron. Naukowcy stworzyli również internetowe repozytorium danych MECP2, portal MECP2-NeuroAtlas, jako źródło informacji dla innych badaczy.
„Myślę, że ten artykuł fundamentalnie zmieni ludzkie rozumienie tego, jak MECP2 powoduje zespół Retta. Mamy zupełnie nowe zrozumienie mechanizmu i może to zapewnić nowe możliwości opracowywania metod leczenia tej choroby” — mówi Janisch, która jest również profesorem biologii w MIT.
Głębsze zrozumienie MECP2 w mózgu
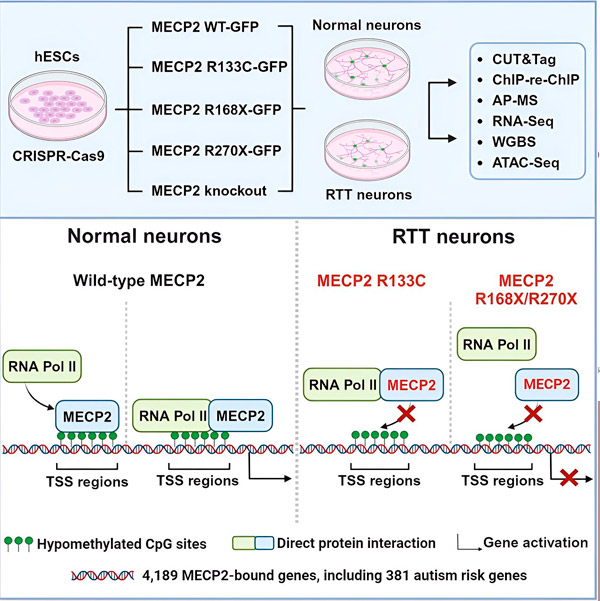
Naukowcy najpierw stworzyli szczegółową mapę, gdzie MECP2 wiąże się w ludzkich sekwencjach genów neuronalnych, albo w obrębie genów, albo w regionach regulacyjnych DNA w ich pobliżu. Zastosowali podejście zwane CUT&Tag, które może precyzyjnie określić interakcje białek z DNA.
Naukowcy odkryli ponad 4000 genów powiązanych z MECP2. Powtórzyli mapowanie w neuronach z powszechnymi mutacjami MECP2 powiązanymi z zespołem Retta, aby określić, gdzie MECP2 jest wyczerpany w stanie chorobowym.
Wiedza o tym, z którymi genami wiąże się MECP2, pozwoliła Liu i Flamierowi rozpocząć tworzenie połączeń między celami MECP2 a zdrowiem mózgu. Odkryli, że wiele z jego celów bierze udział w rozwoju i funkcjonowaniu aksonów i synaps neuronalnych.
Porównali również swoją listę celów MECP2 z bazą danych Simons Foundation Autism Research Initiative (SFARI) zawierającą geny związane z autyzmem i odkryli, że 381 genów w tej bazie danych to cele MECP2.
Źródło: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Wyniki te mogą pomóc wyjaśnić mechanizmy leżące u podstaw objawów autyzmu w zespole Retta i stanowić dobry punkt wyjścia do zbadania możliwej roli MECP2 w autyzmie.
„Stworzyliśmy pierwszą zintegrowaną mapę epigenomu MECP2 w zdrowiu i chorobie, a mapa ta może pokierować przyszłymi badaniami” — mówi Liu. „Wiedza o tym, które geny są celami MECP2, a które są bezpośrednio zakłócane w chorobie, zapewnia solidne podstawy do zrozumienia zespołu Retta i zadawania pytań o regulację genów w neuronach”.
Naukowcy przyjrzeli się również temu, czy MECP2 zwiększał, czy zmniejszał ekspresję swoich genów docelowych. Zgodnie z historią, w której MECP2 był identyfikowany przez niektórych jako aktywator, a przez innych jako represor, Liu i Flamier znaleźli przykłady, w których MECP2 odgrywał obie role.
Jednakże, podczas gdy MECP2 jest częściej postrzegany jako represor, Liu i Flamier odkryli, że jest on głównie aktywatorem — potwierdzając wcześniejsze ustalenia Jaenischa i Liu. Jeden nowy eksperyment wykazał, że MECP2 aktywuje co najmniej 80% swoich celów, a inny wykazał, że aktywuje do 88% swoich celów.
Mapa genów docelowych stworzona przez badaczy dostarczyła dodatkowych informacji na temat roli MECP2 jako aktywatora. Odkryli, że w przypadku genów, które MECP2 aktywuje, zazwyczaj wiąże się z regionem DNA znajdującym się przed genem, zwanym miejscem rozpoczęcia transkrypcji.
To miejsce, w którym maszyneria komórkowa inicjuje proces transkrypcji genu do RNA, po czym RNA jest tłumaczone na funkcjonalne białko, które jest produktem ekspresji genu. Obecność MECP2 w miejscu rozpoczęcia transkrypcji, gdzie rozpoczyna się ekspresja genu, jest zgodna z jego rolą jako aktywatora genu.
Następnie naukowcy postanowili ustalić, jaką rolę odgrywa MECP2 w aktywacji genów. Przyjrzeli się cząsteczkom, z którymi MECP2 wiąże się w tym miejscu, oprócz DNA, i odkryli, że MECP2 oddziałuje bezpośrednio z kompleksem białkowym zwanym polimerazą RNA II (RNA Pol II). RNA Pol II to kluczowa maszyna komórkowa, która przepisuje DNA na RNA. RNA Pol II nie może samodzielnie znaleźć genów, więc wymaga różnych kofaktorów, czyli współpracowników białkowych, aby pomóc jej wykonać swoje zadanie.
Naukowcy sugerują, że MECP2 służy jako jeden z takich kofaktorów, pomagając RNA Pol II zainicjować transkrypcję w genach, w których wiąże się MECP2. Analiza strukturalna MECP2 zidentyfikowała części cząsteczki, które wiążą się z RNA Pol II, a inne eksperymenty potwierdziły, że utrata MECP2 zmniejsza obecność RNA Pol II w odpowiednich miejscach rozpoczęcia transkrypcji, a także poziomy ekspresji genów docelowych.
Sugeruje to, że zespół Retta może być spowodowany zmniejszoną transkrypcją genów docelowych dla MECP2 z powodu mutacji MECP2, które uniemożliwiają mu wiązanie się z RNA Pol II lub wiązanie się z DNA. Zgodnie z tą ideą, najczęstsze mutacje MECP2 związane z chorobą to skrócenia: mutacje, w których brakuje części białka, co może zmienić interakcję między MECP2 a RNA Pol II.
Naukowcy mają nadzieję, że ich odkrycia nie tylko zmienią naszą wiedzę na temat MECP2, ale że głębsze i szersze zrozumienie wpływu MECP2 na rozwój i funkcjonowanie mózgu może doprowadzić do nowych spostrzeżeń, które pomogą osobom z zespołem Retta i pokrewnymi zaburzeniami, w tym autyzmem.
„Ten projekt jest świetnym przykładem współpracy w laboratorium Janisch” — mówi Flamier. „Rudolf i ja mieliśmy konkretny problem związany z zespołem Retta, a ja miałem doświadczenie z technologią CUT&Tag, która mogła rozwiązać ten problem. Dzięki dyskusji zdaliśmy sobie sprawę, że możemy połączyć nasze wysiłki i teraz mamy świetne repozytorium informacji o MECP2 i jego powiązaniach z chorobami”.