Ekspert medyczny artykułu

Nowe publikacje
Zespół Pierre'a Robina
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.

Zespół Pierre’a Robina, znany również w medycynie jako anomalia Robina, jest wrodzoną patologią rozwoju części szczękowej twarzy. Choroba otrzymała swoją nazwę na cześć francuskiego dentysty P. Robina, który jako pierwszy opisał wszystkie jej objawy. Lannelongue i Menard po raz pierwszy opisali zespół Pierre’a Robina w 1891 r. w swoim raporcie dotyczącym dwóch pacjentów z mikrognacją, rozszczepem podniebienia i retroglossoptozą. W 1926 r. Pierre-Robin opublikował przypadek choroby u niemowlęcia z objawami klasycznego zespołu. Do 1974 r. triada objawów była znana jako zespół Robina-Pierre’a. Jednak zespół ten jest obecnie używany do opisu wad rozwojowych z jednoczesną obecnością wielu anomalii.
Epidemiologia
Jest to heterogenna wrodzona wada, której częstość występowania wynosi 1 na 8500 żywych urodzeń. Stosunek mężczyzn do kobiet wynosi 1:1, z wyjątkiem postaci sprzężonej z chromosomem X.
Wśród tych pacjentów 50% niemowląt ma niepełny rozszczep podniebienia miękkiego, reszta rodzi się z wysklepionym i niezwykle wysokim podniebieniem, ale bez rozszczepu.
Przyczyny Zespół Pierre'a Robina
Rozważana jest możliwość autosomalnego recesywnego dziedziczenia choroby. Istnieją dwa typy zespołu w zależności od etiologii: izolowany i genetycznie uwarunkowany. Typ izolowany rozwija się z powodu ucisku dolnej części szczęki w trakcie rozwoju embrionalnego. Ucisk może rozwinąć się z powodu:
- Obecność zlokalizowanych ognisk zapalnych w macicy (torbiele, blizny, guzy).
- Ciąża mnoga.
Ponadto rozwój szczęki u płodu może zostać zaburzony przez:
- Zakażenia wirusowe, na które przyszła matka cierpiała w czasie ciąży.
- Zaburzenia neurotroficzne.
- Niedostateczna ilość kwasu foliowego w organizmie kobiety ciężarnej.
Patogeneza
Zespół Pierre’a Robina jest spowodowany zaburzeniami embrionalnymi, które powstają w wyniku szerokiego zakresu patologii w okresie prenatalnym.
Istnieją trzy teorie patofizjologiczne, które mogą wyjaśniać występowanie zespołu Pierre’a Robina.
Teoria mechaniczna: Ta teoria jest najbardziej prawdopodobna. Niedorozwój aparatu żuchwowego występuje między 7. a 11. tygodniem ciąży. Wysokie położenie języka w jamie ustnej prowadzi do powstania rozszczepów podniebienia, przez co żyła główna nie zamyka się. Ta teoria wyjaśnia klasyczny rozszczep w kształcie odwróconej litery U i brak towarzyszącego mu rozszczepu wargi. Małowodzie może odgrywać rolę w etiologii, ponieważ brak płynu owodniowego może prowadzić do deformacji brody i późniejszego ucisku języka między żyłą główną.
Teoria neurologiczna: Opóźnienie rozwoju neurologicznego zaobserwowano w elektromiografii mięśni języczka i strun gardłowych, a także w rozwoju smaku z powodu opóźnienia przewodzenia w nerwie podjęzykowym.
Teoria dysneuroregulacji rombencefalonu: Teoria ta opiera się na zakłóceniu rozwoju rombencefalonu w ontogenezie.
Niedostateczny rozwój dolnej części szczęki dziecka prowadzi do znacznego zmniejszenia jamy ustnej. To z kolei powoduje tzw. pseudomakroglossię, czyli przemieszczenie języka na tył ściany gardła. Ta patologia prowadzi do rozwoju niedrożności dróg oddechowych.
Dopóki dziecko płacze lub się rusza, drogi oddechowe pozostają drożne, ale gdy tylko dziecko zaśnie, niedrożność znów się pojawia.
Z powodu zaburzeń oddechowych proces karmienia dziecka jest bardzo utrudniony. W tym czasie prawie zawsze dochodzi do niedrożności dróg oddechowych. Jeśli nie zostanie zastosowana korekcja medyczna, taka patologia może doprowadzić do poważnego wyczerpania całego organizmu, a nawet śmierci.
Objawy Zespół Pierre'a Robina
Chorobę charakteryzują trzy główne objawy:
- Mikrognacja dolna (niedorozwój żuchwy, występuje w 91,7% przypadków choroby). Charakteryzuje się cofnięciem dolnego łuku zębowego o 10-12 mm za łuk górny. Żuchwa ma mały trzon, kąt rozwarty. Dziecko osiąga prawidłowy rozwój około 5-6 roku życia.
- Glossoptoza (cofnięcie języka z powodu jego niedostatecznego rozwoju, obserwowane w 70-85% przypadków).
- Makroglossia i ankyloglossia są stosunkowo rzadkimi objawami, obserwowanymi w 10-15% przypadków.
- Na niebie pojawia się pęknięcie.
- Bradypnoe i duszność.
- Łagodna sinica.
- Uduszenie, do którego najczęściej dochodzi podczas próby karmienia dziecka.
- Połknięcie jest niemożliwe lub bardzo utrudnione.
- Mam ochotę wymiotować.
- Anomalie małżowiny usznej w 75% przypadków.
- Niedosłuch przewodzeniowy występuje u 60% chorych, natomiast zarośnięcie przewodu słuchowego zewnętrznego występuje zaledwie u 5% chorych, niedostateczna pneumatyzacja jamy sutkowej kości skroniowej.
- Anomalie ucha wewnętrznego (aplazja kanałów półkolistych bocznych, wielkiego wodociągu przedsionkowego, zanik komórek włoskowatych ślimaka).
- Wady rozwojowe nosa zdarzają się rzadko i polegają głównie na nieprawidłowościach nasady nosa.
- Wady zębowe występują w 30% przypadków. Laryngomalacja i niewydolność podniebienno-gardłowa występują u około 10-15% pacjentów z zespołem Pierre’a Robina.
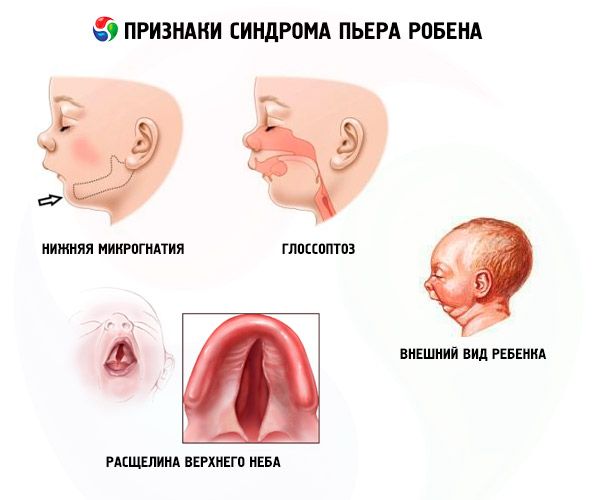
Objawy systemowe zespołu Pierre’a Robina
W 10-85% zarejestrowanych przypadków opisano systemowe anomalie rozwojowe.
Nieprawidłowości oka występują u 10-30% pacjentów. Mogą obejmować: nadwzroczność, krótkowzroczność, astygmatyzm, stwardnienie rogówki i zwężenie przewodu nosowo-łzowego.
Patologie układu sercowo-naczyniowego: łagodne szmery serca, zwężenie tętnicy płucnej, przetrwały przewód tętniczy, okienko owalne, ubytek przegrody międzyprzedsionkowej i nadciśnienie płucne. Ich częstość występowania waha się od 5 do 58%.
Anomalie związane z układem mięśniowo-szkieletowym (70-80% przypadków): syndaktylia, dysplastyczne paliczki, polidaktylia, klinodaktylia, hipermobilność stawów i oligodaktylia kończyn górnych. Anomalie kończyn dolnych: anomalie stóp (stopa końsko-szpotawa, przywodzenie kości śródstopia), wady rozwojowe kości udowej (koślawość lub szpotawość miednicy, krótkie kości udowe), anomalie bioder (wrodzone zwichnięcia, przykurcze), anomalie stawu kolanowego (GENU VALGUS, synchondrosis). Wady rozwojowe kręgosłupa: skolioza, kifoza, lordoza, dysplazja kręgów, agenezja kości krzyżowej i zatoki guzicznej.
Patologia ośrodkowego układu nerwowego: padaczka, opóźnienia w rozwoju układu nerwowego, wodogłowie. Częstotliwość występowania wad ośrodkowego układu nerwowego wynosi około 50%.
Anomalie układu moczowo-płciowego: niezstąpione jądra (25%), wodonercze (15%) i wodniak jądra (10%).
Powiązane zespoły i schorzenia: zespół Sticklera, zespół trisomii 11q, trisomia 18, zespół delecji 4q, reumatoidalne zapalenie stawów, hipochondroplazja, zespół Moebiusa.
Gradacja
Wyróżnia się trzy stopnie nasilenia choroby, które zależą od stanu dróg oddechowych dziecka:
- Łagodne - występują drobne problemy z karmieniem, ale oddychanie jest prawie nieskomplikowane. Leczenie odbywa się ambulatoryjnie.
- Umiarkowane – oddychanie jest umiarkowanie utrudnione, karmienie dziecka jest umiarkowanie utrudnione. Leczenie odbywa się w szpitalu.
- Ciężki – oddychanie jest bardzo utrudnione, dziecko nie może być karmione normalnie. Konieczne jest użycie specjalnych urządzeń (sonda donosowa).
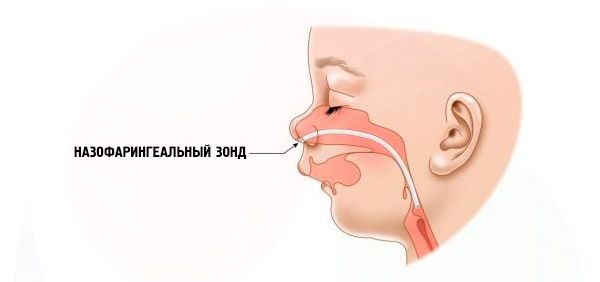
Komplikacje i konsekwencje
Połączenie mikrognacji i opadnięcia języka może prowadzić do poważnych powikłań oddechowych oraz problemów z karmieniem dziecka.
Zespół Pierre’a Robina powoduje następujące powikłania:
- Oddychanie stridozą z powodu niedrożności dróg oddechowych. Laryngomalacja lub nawet asfiksja senna.
- Rozwój psychomotoryczny dziecka znacznie odbiega od rozwoju jego rówieśników.
- Rozwój fizyczny również pozostaje w tyle.
- U pacjentów występuje upośledzenie mowy.
- Częste infekcje ucha, które stają się przewlekłe i prowadzą do upośledzenia słuchu.
- W przypadku zespołu obturacyjnego bezdechu sennego częstość występowania zgonów podczas snu waha się w 14-91% przypadków.
- Problemy z zębami.
Diagnostyka Zespół Pierre'a Robina
Diagnoza zespołu Pierre’a Robina nie jest trudna. Opiera się na objawach klinicznych. Aby wykluczyć inne patologie, bardzo ważne jest skonsultowanie się z genetykiem.
Dzieci z wrodzoną anomalią Robina mają problemy z oddychaniem od urodzenia z powodu ciągłego zapadania się języka. Niemowlę jest niespokojne, jego skóra jest sina, z klatki piersiowej wydobywa się świszczący oddech podczas wdechu. Podczas karmienia może wystąpić zakrztuszenie. Diagnozę można również postawić na podstawie nietypowego wyglądu dziecka - „ptasiej twarzy”. Często u pacjentów rozwijają się inne wady: krótkowzroczność, zaćma, patologia układu moczowo-płciowego, patologia serca, anomalie w rozwoju kręgosłupa.
Na podstawie tych objawów klinicznych specjalista bez trudu postawi prawidłową diagnozę.
Z kim się skontaktować?
Leczenie Zespół Pierre'a Robina
Leczenie przeprowadza się bezpośrednio po urodzeniu dziecka z zespołem Pierre’a Robina. Jeśli choroba ma łagodny przebieg, to w celu poprawy stanu pacjenta konieczne jest ciągłe trzymanie dziecka w pozycji pionowej lub leżenie na brzuszku. Główka dziecka powinna być pochylona do klatki piersiowej. Podczas karmienia nie zaleca się trzymania dziecka w pozycji poziomej, aby pokarm nie dostał się do dróg oddechowych.
Jeśli niedorozwój żuchwy jest bardzo wyraźny, stosuje się interwencję chirurgiczną, aby przywrócić cofnięty język do normalnej fizjologicznej pozycji. W ciężkich przypadkach język jest podciągany i mocowany na dolnej wardze. W bardzo ciężkich przypadkach należy wykonać tracheostomię, glossopeksję i dystrakcyjną osteogenezę żuchwy.
Stosuje się również leczenie konserwatywne.
Leki
Fenobarbital. Środek nasenny i uspokajający, ma działanie przeciwdrgawkowe. Każda tabletka zawiera 100 ml fenobarbitalu. Dawkowanie jest indywidualne, ponieważ zależy od ciężkości choroby i stanu dziecka. Lek jest zabroniony dla pacjentów z niewydolnością wątroby, hiperkinezą, anemią, miastenią, porfirią, cukrzycą, depresją i nietolerancją składników. Podczas przyjmowania leku możliwe są następujące objawy: zawroty głowy, osłabienie, halucynacje, agranulocytoza, nudności, niskie ciśnienie krwi i alergie.
Klonazepam. Lek przepisywany w leczeniu padaczki. Lek zawiera substancję czynną klonazepam, która jest pochodną benzodiazepiny. Ma działanie przeciwdrgawkowe, przeciwlękowe i zwiotczające mięśnie. Dawkę ustala lekarz prowadzący, ale nie powinna przekraczać maksymalnej dawki - 250 mcg na dobę. Nie należy przyjmować w przypadku bezsenności, hipertonii mięśniowej, pobudzenia psychoruchowego, napadów paniki. Podczas przyjmowania leku możliwe są następujące objawy: letarg, nudności, bolesne miesiączkowanie, ból głowy, leukopenia, zatrzymanie lub nietrzymanie moczu, łysienie, alergia.
Sibazon. Dostępny w postaci roztworu i tabletek doodbytniczych. Substancją czynną jest pochodna benzodiazepiny (sibazon). Ma działanie uspokajające, przeciwlękowe, przeciwdrgawkowe. Dawkowanie ustalane jest indywidualnie. Pacjenci z przewlekłą hiperkapnią, miastenią, nietolerancją benzodiazepin nie powinni przyjmować leku. Podczas stosowania leku mogą wystąpić następujące objawy: nudności, zaparcia, ból głowy, zawroty głowy, czkawka, nietrzymanie moczu, alergie.
Liofilizat Cortexin. Lek o działaniu nootropowym. Lek zawiera kompleks rozpuszczalnych w wodzie frakcji polipeptydowych i glicyny. Dawkowanie jest indywidualne i jest przepisywane przez lekarza prowadzącego zgodnie ze stanem pacjenta. Pacjenci z nietolerancją cortexinu nie mogą przyjmować leku. Lek może powodować reakcje alergiczne.
Leczenie fizjoterapeutyczne
Zazwyczaj w łagodnych stadiach zespołu stosuje się terapię ułożeniową, polegającą na ułożeniu dziecka na brzuchu w pozycji pionowej, aż do momentu, gdy siła grawitacji zmusi żuchwę do prawidłowego rozwoju.
Leczenie chirurgiczne
Leczenie chirurgiczne jest stosowane przede wszystkim w celu skorygowania glossoptosis. Istnieje kilka metod:
- Podparcie języka srebrną nicią. Nić przechodzi przez dolną część dziąsła i dolną wargę. Metoda ta nazywa się Douglas.
- Metoda Duhamela - gruba srebrna nić jest przeciągana przez podstawę języka pacjenta i oba policzki. Stosować nie dłużej niż trzydzieści dni.
- Urządzenia ortopedyczne do prostowania i unieruchamiania języka.
- W wieku jednego roku można przeprowadzić operację korygującą rozszczep podniebienia.
Prognoza
Rokowanie i przebieg choroby są poważne. Najczęściej śmierć następuje w pierwszych dniach życia w umiarkowanym i ciężkim stadium choroby (przyczyną jest uduszenie). Ponadto ryzyko zgonu w pierwszym roku jest dość wysokie ze względu na liczne infekcje.
W przypadku pacjentów powyżej drugiego roku życia rokowanie jest pomyślne.
 [ 36 ]
[ 36 ]