Ekspert medyczny artykułu

Nowe publikacje
Zespół Ushera
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Zespół Ushera to choroba dziedziczna, która objawia się całkowitą głuchotą od urodzenia, a także postępującą ślepotą z wiekiem. Utrata wzroku jest związana z retinitis pigmentosa, procesem zwyrodnienia barwnikowego siatkówki. Wiele osób z zespołem Ushera ma również poważne problemy z równowagą.
Epidemiologia
Dzięki badaniom udało się ustalić, że zespół Ushera dotyczy około 8% badanych dzieci głuchoniemych (badania przeprowadzono w specjalnych placówkach dla osób głuchoniemych). Zapalenie barwnikowe siatkówki obserwowano u 6-10% pacjentów cierpiących na wrodzoną głuchotę, która z kolei występuje u około 30% osób z chorobą barwnikową siatkówki.
Uważa się, że choroba ta występuje u około 3-10 osób na 100 tysięcy na świecie. Występuje w równym stopniu u kobiet i mężczyzn. Około 5-6% światowej populacji cierpi na ten zespół. Około 10% wszystkich przypadków głębokiej głuchoty u dzieci występuje z powodu zespołu Ushera typu I, jak również typu II.
W Stanach Zjednoczonych typy 1 i 2 są najczęstszymi typami. Razem stanowią około 90 do 95 procent wszystkich przypadków zespołu Ushera u dzieci.
Przyczyny Zespół Ushera
Zespół Ushera typu I, II i III ma przyczynę autosomalną recesywną, podczas gdy typ IV jest uważany za zaburzenie chromosomu X. Przyczyny ślepoty i głuchoty występujące w przypadku tego zespołu nie zostały jeszcze wystarczająco zbadane. Zakłada się, że osoby z tą chorobą są nadwrażliwe na składniki, które mogą uszkodzić strukturę DNA. Ponadto choroba ta może być związana z zaburzeniami układu odpornościowego, ale w tym przypadku nie ma dokładnego obrazu tego procesu.
W 1989 r. po raz pierwszy zidentyfikowano nieprawidłowości chromosomalne u pacjentów z chorobą typu II, co w przyszłości może doprowadzić do wyizolowania genów powodujących zespół. Możliwe jest również zidentyfikowanie tych genów u nosicieli i opracowanie specjalnych prenatalnych testów genetycznych.
 [ 8 ]
[ 8 ]
Czynniki ryzyka
Zespół ten dziedziczy się, gdy oboje rodzice są dotknięci chorobą, tj. dziedziczy się go w sposób recesywny. Dziecko może również odziedziczyć chorobę, jeśli jego rodzice są nosicielami genu. Jeśli oboje przyszli rodzice mają ten gen, prawdopodobieństwo urodzenia dziecka z tym zespołem wynosi 1 do 4. Osoba, która ma tylko jeden gen dla zespołu, jest uważana za nosiciela, ale nie ma objawów zaburzenia. Obecnie nie jest jeszcze możliwe ustalenie, czy dana osoba ma gen dla tej choroby.
Jeśli dziecko urodzi się, gdy jedno z rodziców nie ma takiego genu, to prawdopodobieństwo, że odziedziczy ono zespół chorobowy jest bardzo małe, ale na pewno będzie nosicielem.
Objawy Zespół Ushera
Objawy zespołu Ushera obejmują utratę słuchu i nieprawidłową akumulację komórek pigmentowych w strukturach oka. Następnie u pacjenta rozwija się zwyrodnienie siatkówki, co powoduje pogorszenie widzenia, a w najcięższych przypadkach nawet jego utratę.
Niedosłuch odbiorczy może być łagodny lub całkowity i zwykle nie postępuje od urodzenia. Jednak choroba barwnikowa siatkówki może zacząć rozwijać się w dzieciństwie lub później. Wyniki badań wykazały, że ostrość wzroku centralnego może być utrzymywana przez wiele lat, nawet gdy pogarsza się widzenie peryferyjne (stan zwany „widzeniem tunelowym”).
Są to główne objawy choroby, którym czasami mogą towarzyszyć inne zaburzenia, takie jak psychoza i inne zaburzenia psychiczne, problemy z uchem wewnętrznym i/lub zaćma.
Formularze
W toku badań wyróżniono 3 typy tej choroby oraz czwartą, dość rzadką postać.
Typ I choroby charakteryzuje się wrodzoną całkowitą głuchotą, a także zaburzeniami równowagi. Często takie dzieci zaczynają chodzić dopiero w wieku 1,5 roku. Pogorszenie widzenia zwykle rozpoczyna się w wieku 10 lat, a ostateczny rozwój stanu kurzej ślepoty rozpoczyna się w wieku 20 lat. U dzieci z tym typem choroby może wystąpić postępujące pogorszenie widzenia peryferyjnego.
W przypadku choroby typu II obserwuje się umiarkowaną lub wrodzoną głuchotę. W tym przypadku pogorszenie częściowej głuchoty często już nie występuje. Zapalenie barwnikowe siatkówki zaczyna rozwijać się pod koniec okresu dojrzewania lub po 20 latach. Rozwój ślepoty nocnej zwykle rozpoczyna się w wieku 29-31 lat. Upośledzenie ostrości wzroku w przypadku patologii typu II postępuje na ogół nieco wolniej niż w typie I.
Typ III choroby charakteryzuje się postępującą utratą słuchu, rozpoczynającą się zwykle w okresie dojrzewania, a także stopniowym rozwojem w tym samym okresie (nieco później niż utrata słuchu) retinitis pigmentosa, która może stać się czynnikiem wpływającym na rozwój postępującej ślepoty.
Objawy patologii typu IV występują głównie u mężczyzn. W tym przypadku obserwuje się również postępujące zaburzenia oraz utratę słuchu i wzroku. Ta forma jest bardzo rzadka i zwykle ma charakter chromosomu X.
Diagnostyka Zespół Ushera
Rozpoznanie zespołu Ushera ustala się na podstawie zaobserwowanego u pacjenta połączenia nagłej głuchoty i postępującej utraty wzroku.
Testy
W celu wykrycia mutacji można zlecić wykonanie specjalnego testu genetycznego.
Odkryto jedenaście loci genetycznych, które mogą powodować rozwój zespołu Ushera, a także zidentyfikowano dziewięć genów, które z całą pewnością są przyczyną tego zaburzenia:
- Typ 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Typ 2: ush2a, VLGR1, WHRN.
- Zespół Ushera typu 3: USH3A.
Naukowcy z NIDCD wraz z kolegami z uniwersytetów w Nowym Jorku i Izraelu zidentyfikowali mutację zwaną R245X w genie Pcdh15, która jest odpowiedzialna za duży odsetek przypadków zespołu Ushera typu 1 wśród populacji żydowskiej.
Aby dowiedzieć się więcej o laboratoriach, które przeprowadzają badania kliniczne, odwiedź stronę https://www.genetests.org i wyszukaj w katalogu laboratoriów hasło „zespół Ushera”.
Aby dowiedzieć się więcej o istniejących badaniach klinicznych, które obejmują testy genetyczne na zespół Ushera, odwiedź stronę https://www.clinicaltrials.gov i wyszukaj „zespół Ushera” lub „testy genetyczne na zespół Ushera”.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnostyka instrumentalna
Istnieje kilka metod diagnostyki instrumentalnej:
- Badanie dna oka w celu wykrycia obecności plam pigmentacyjnych na siatkówce, a także zwężenia naczyń siatkówki;
- Elektroretinogram, który pozwala wykryć początkowe degeneracyjne odchylenia siatkówki oka. Pokazuje wygaśnięcie ścieżek elektroradiograficznych;
- Elektronystagmogram (ENG) pozwala na pomiar mimowolnych ruchów gałek ocznych, które mogą wskazywać na obecność braku równowagi.
- Audiometria, która służy określeniu obecności głuchoty i jej stopnia zaawansowania.
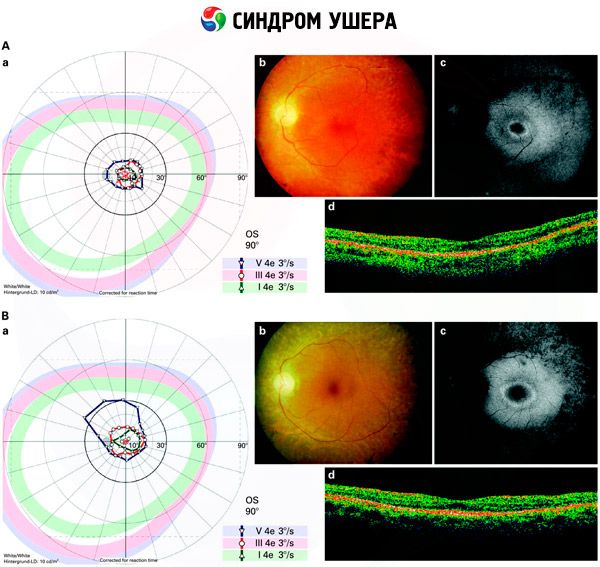
Diagnostyka różnicowa
Zespół Ushera należy odróżnić od innych podobnych zaburzeń.
Zespół Hallgrena, który charakteryzuje się wrodzoną utratą słuchu i postępującą utratą wzroku (rozwija się również zaćma i oczopląs). Dodatkowe objawy obejmują ataksję, zaburzenia psychomotoryczne, psychozę i upośledzenie umysłowe.
Zespół Alströma, który jest chorobą dziedziczną, w której siatkówka ulega zwyrodnieniu, powodując utratę widzenia centralnego. Zespół ten jest związany z otyłością dziecięcą. Jednocześnie po 10 latach zaczynają rozwijać się cukrzyca i utrata słuchu.
Różyczka u kobiety w ciąży w pierwszym trymestrze może powodować różne nieprawidłowości w rozwoju dziecka. Wśród konsekwencji takiej nieprawidłowości są utrata słuchu, a także (lub) problemy ze wzrokiem, a oprócz tego różne wady rozwojowe.
Z kim się skontaktować?
Leczenie Zespół Ushera
Obecnie nie ma lekarstwa na zespół Ushera. Dlatego terapia w tym przypadku polega głównie na spowolnieniu procesu utraty wzroku, a także na kompensacji utraty słuchu. Możliwe metody leczenia obejmują:
- Przyjmowanie witaminy A (niektórzy okuliści uważają, że duże dawki palmitynianu witaminy A mogą spowolnić, ale nie zatrzymać rozwój retinitis pigmentosa);
- Wszczepienie pacjentowi specjalnych urządzeń elektronicznych (aparatów słuchowych, implantów ślimakowych).
Okuliści zalecają, aby większość dorosłych z typowymi formami retinitis pigmentosa przyjmowała 15 000 IU (jednostek międzynarodowych) palmitynianu witaminy A dziennie pod nadzorem. Ponieważ osoby z zespołem Ushera typu 1 nie zostały uwzględnione w badaniu, nie zaleca się dużych dawek witaminy A dla tej grupy pacjentów. Osoby, które rozważają przyjmowanie witaminy A, powinny omówić tę opcję leczenia ze swoim lekarzem. Inne zalecenia dotyczące tej opcji leczenia obejmują:
- Zmiana diety i włączenie do niej produktów bogatych w witaminę A.
- Kobiety planujące ciążę powinny zaprzestać przyjmowania dużych dawek witaminy A na trzy miesiące przed planowanym poczęciem ze względu na zwiększone ryzyko wystąpienia wad wrodzonych.
- Kobiety w ciąży nie powinny przyjmować dużych dawek witaminy A ze względu na zwiększone ryzyko wystąpienia wad wrodzonych.
Ważne jest również przystosowanie takiego dziecka do życia społecznego. Wymaga to pomocy nauczycieli edukacji specjalnej i psychologów. W przypadku, gdy pacjent zaczął doświadczać postępującej utraty wzroku, należy nauczyć go posługiwania się językiem migowym.
Prognoza
Zespół Ushera ma niekorzystne rokowanie. Pole widzenia i jego ostrość zaczynają się pogarszać w okresie 20-30 lat u większości pacjentów z tą chorobą dowolnego typu. W niektórych przypadkach dochodzi do całkowitej obustronnej utraty wzroku. Utrata słuchu, której zawsze towarzyszy niemota, bardzo szybko rozwija się do całkowitej obustronnej utraty słuchu.