Ekspert medyczny artykułu

Nowe publikacje
Podostra martwicza encefalomiopatia Leaha
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Choroba została po raz pierwszy wspomniana w 1951 r. Do tej pory opisano ponad 120 przypadków. Choroba Leigha (OMIM 256000) jest genetycznie heterogeniczną chorobą, która może być dziedziczona albo jądrowo (autosomalnie recesywnie lub sprzężona z chromosomem X), albo mitochondrialnie (rzadziej).
 [
[ Przyczyny zespołu Leah
Choroba opiera się na niedoborze enzymów, które zapewniają produkcję energii, głównie z powodu zaburzenia metabolizmu kwasu pirogronowego i wady transportu elektronów w łańcuchu oddechowym. Rozwija się niedobór kompleksu dehydrogenazy pirogronianowej (podjednostka a-E1), karboksylazy pirogronianowej, kompleksu 1 (reduktaza NAD-koenzymu Q) i kompleksu 4 (oksydaza cytochromowa) łańcucha oddechowego.
Ustalono, że defekty karboksylazy pirogronianowej, kompleksu 1 (NAD-koenzym Q-reduktaza) i kompleksu 4 (oksydaza cytochromowa) łańcucha oddechowego dziedziczą się w sposób autosomalny recesywny, defekty kompleksu dehydrogenazy pirogronianowej (podjednostka a-E1) dziedziczą się w sposób recesywny sprzężony z chromosomem X. W przypadku mutacji punktowych mtDNA, które dotyczą 6. podjednostki ATPazy, typowe jest dziedziczenie mitochondrialne. Najczęściej występuje mutacja miscens, związana z zastąpieniem tyminy guaniną lub cytozyną w pozycji 8993 mtDNA. Rzadziej występuje mutacja w pozycji 9176 mtDNA. Ze względu na fakt, że mutacja T8993G jest głównym defektem w zespole NARP, opisano rodziny z tymi dwiema chorobami. U dzieci opisano również mutację w mtDNA w pozycji 8344, która występuje w zespole MERRF.
Zakłada się, że w przypadku akumulacji zmutowanego mtDNA w większości mitochondriów rozwija się ciężki przebieg zespołu Leigha. W mitochondrialnej genezie tego schorzenia zmutowane mtDNA występuje w 90% wszystkich mitochondriów. Patogeneza wiąże się z zaburzeniem wytwarzania energii w komórkach i rozwojem kwasicy mleczanowej.
Objawy zespołu Leah
Pierwsze objawy choroby pojawiają się w młodym wieku (1-3 lata). Znane są jednak przypadki ujawnienia się choroby w wieku 2 tygodni i 6-7 lat. Początkowo rozwijają się niespecyficzne zaburzenia: opóźniony rozwój psychoruchowy, zmniejszony apetyt, epizody wymiotów, niedobór masy ciała. Następnie nasilają się objawy neurologiczne: hipotonia mięśniowa lub dystonia z przejściem w hipertonię, napady mioklonii lub napady toniczno-kloniczne, drżenie kończyn, choreoatetoza, zaburzenia koordynacji, osłabienie odruchów ścięgnistych, letarg, senność. Neurodegeneracja mózgu postępuje. Nasilają się objawy niewydolności piramidowej i pozapiramidowej, upośledzony jest akt połykania. Często obserwuje się takie zmiany w narządzie wzroku, jak opadanie powieki, oftalmoplegia, zanik nerwów wzrokowych, rzadziej zwyrodnienie barwnikowe siatkówki. Czasami rozwija się kardiomiopatia przerostowa, pojawiają się epizody tachypnoe.
Rzadko choroba przebiega jako ostra encefalopatia. Bardziej typowy jest przebieg przewlekły lub podostry, który prowadzi do zgonu kilka lat po wystąpieniu choroby. Przy szybkim przebiegu (kilka tygodni) śmierć następuje w wyniku porażenia ośrodka oddechowego.
Diagnostyka zespołu Leah
Biochemiczne badanie krwi ujawnia kwasicę mleczanową z powodu gromadzenia się kwasu mlekowego i pirogronowego we krwi i płynie mózgowo-rdzeniowym, a także wzrostu zawartości alaniny we krwi. Może być również podwyższony poziom ciał ketonowych. W moczu wykrywa się zwiększone wydalanie kwasów organicznych: mlekowego, fumarowego itp. Często obniża się poziom karnityny we krwi i tkankach.
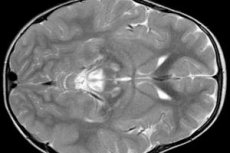
Wyniki EEG ujawniają ogniskowe oznaki aktywności padaczkowej. Dane MRI ujawniają powiększenie komór mózgowych, obustronne uszkodzenie mózgu, zwapnienie jąder podstawy (jądra ogoniastego, skorupy, istoty czarnej, gałki bladej). Można również wykryć zanik półkul mózgowych i materii mózgowej.
Badanie morfologiczne ujawnia duże zmiany w materii mózgowej: symetryczne ogniska martwicy, demielinizacji i gąbczastego zwyrodnienia mózgu, głównie jego części środkowej, mostu, jąder podstawy, wzgórza i nerwu wzrokowego. Obraz histologiczny obejmuje torbielowate zwyrodnienie tkanki mózgowej, gliozę astrocytarną, obumieranie neuronów i wzrost liczby mitochondriów w komórkach. W mięśniach szkieletowych występuje gromadzenie się inkluzji lipidowych, spadek reakcji histochemicznej na kompleksy 1 i 4 łańcucha oddechowego, podsarkolemmalne gromadzenie się mitochondriów, nieprawidłowe mitochondria z dezorganizacją grzebieni. Zjawisko RRF często nie jest wykrywane.
Jak zbadać?
Jakie testy są potrzebne?
Использованная литература