Ekspert medyczny artykułu

Nowe publikacje
Postępujący paraliż: formy kliniczne, stadia i przebieg
Ostatnia aktualizacja: 27.10.2025

Stosujemy ścisłe wytyczne dotyczące źródeł i linkujemy wyłącznie do renomowanych stron medycznych, placówek badawczych oraz, w miarę możliwości, do badań recenzowanych przez specjalistów medycznych. Należy pamiętać, że liczby w nawiasach ([1], [2] itd.) to klikalne linki do tych badań.
Jeśli uważasz, że którakolwiek z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, zaznacz ją i naciśnij Ctrl + Enter.
Postępujący paraliż to późna, objawowa postać kiły układu nerwowego, rozwijająca się wiele lat po zakażeniu Treponema pallidum. Atakuje on przede wszystkim korę mózgową, prowadząc do zmian osobowości, upośledzenia pamięci, osłabienia zdolności oceny sytuacji i postępującej demencji. Rozwijają się również dyzartria, drżenie, hiperrefleksja, napady padaczkowe, a czasami niedowład. Bez leczenia choroba stale postępuje. [1]
Historycznie, postępujący paraliż był uważany za główną przyczynę demencji u osób w średnim wieku, przed erą penicyliny. Obecnie występuje rzadziej, ale ze względu na globalny wzrost zachorowań na kiłę, przypadki kiły układu nerwowego, w tym późnej, ponownie stają się coraz częstsze. Wymaga to od lekarzy zachowania szczególnej czujności, ponieważ kiła układu nerwowego może maskować się pod postacią zaburzeń psychicznych, choroby Alzheimera, otępienia czołowo-skroniowego i stwardnienia rozsianego. [2]
Co istotne, postępujący paraliż można leczyć, jeśli diagnoza zostanie postawiona wcześnie i zalecona zostanie odpowiednia terapia penicyliną, a następnie prowadzona będzie obserwacja z monitorowaniem płynu mózgowo-rdzeniowego. Nawet w przypadku późnego wykrycia postęp choroby można zatrzymać, a funkcje poznawcze częściowo poprawić. [3]
Kod według ICD-10 i ICD-11
W ICD-10 postępujący paraliż jest kodowany jako A52.17 „Uogólniony niedowład / Otępienie porażenne” (sekcja „Kiła późna z zajęciem układu nerwowego”). Powiązane kody: A52.11 „Tabes dorsalis”, A52.19 „Inna objawowa kiła układu nerwowego”, A52.2 „Bezobjawowa kiła układu nerwowego”, A52.3 „Kiła układu nerwowego, nieokreślona”. Wybór kodu zależy od obrazu klinicznego i potwierdzenia uszkodzenia ośrodkowego układu nerwowego. [4]
W ICD-11 kiła układu nerwowego jest klasyfikowana jako osobny blok 1A62.0 „Kiła układu nerwowego” z następującymi szczegółami: 1A62.00 „Kiła układu nerwowego bezobjawowa”, 1A62.01 „Kiła układu nerwowego objawowa późna”, 1A62.0Z „Kiła układu nerwowego nieokreślona”. Klinicznie postępujący paraliż odpowiada objawowej późnej kile układu nerwowego. [5]
Tabela 1. Jak kodować postępujący paraliż
| Klasyfikator | Rozdział | Kod | Co to znaczy? |
|---|---|---|---|
| ICD-10 | A52 „Kiła późna” | A52.17 | Postępujący paraliż (demencja paralityczna) |
| ICD-10 | A52 | A52.11 / A52.19 / A52.3 | Kiła grzbietowa / Inne objawowe / Nieokreślona kiła układu nerwowego |
| ICD-11 | 1A62.0 | 1A62.01 | Objawowa późna kiła układu nerwowego (obejmuje postępujący paraliż) |
| ICD-11 | 1A62.0 | 1A62.00 / 1A62.0Z | Kiła układu nerwowego bezobjawowa/nieokreślona |
Epidemiologia
Kiła układu nerwowego rozwija się w każdym stadium kiły, ale klinicznie widoczne późne formy (w tym postępujący paraliż i wklęsłość grzbietu) są charakterystyczne dla nieleczonej lub niewłaściwie leczonej kiły po latach i dekadach od zakażenia. W kontekście globalnego wzrostu zachorowań na kiłę w latach 2010–2020, przypadki kiły układu nerwowego ponownie stały się częstsze; w niektórych populacjach częstość występowania wczesnej kiły układu nerwowego u pacjentów z kiłą sięga 2–5%, podczas gdy późne formy są niższe, ale dane różnią się ze względu na metody rejestracji i dostępność terapii. [6]
Według badań ankietowych i statystyk krajowych, w 2023 roku w Stanach Zjednoczonych odnotowano ponad 200 000 przypadków kiły (w tym wrodzonej), co stanowi najwyższą liczbę od dziesięcioleci. Przy takim wzroście spodziewany jest wzrost całkowitej liczby przypadków kiły układu nerwowego. Dokładne wskaźniki postępującego paraliżu wśród wszystkich postaci kiły układu nerwowego różnią się w zależności od regionu i dostępności badań diagnostycznych. [7]
Tabela 2. Co wpływa na częstość występowania paraliżu postępującego obecnie
| Czynnik | Wpływ |
|---|---|
| Wzrost zachorowań na kiłę w populacji | Zwiększa całkowitą liczbę przypadków kiły układu nerwowego |
| Opóźnienie w diagnozie/terapii | Zwiększa ryzyko późnych form |
| Współinfekcje i niedobory odporności | Przyspieszenie uszkodzeń ośrodkowego układu nerwowego |
| Dostępność penicyliny i nadzór | Zmniejsza występowanie późnych form |
Powody
Przyczyną jest infekcja krętkiem bladym (Treponema pallidum), która nieleczona lub nieleczona utrzymuje się w ośrodkowym układzie nerwowym. Postępujący paraliż jest wynikiem przewlekłej kiły miąższowej układu nerwowego, charakteryzującej się rozproszonym stanem zapalnym, reakcją komórek glejowych i zwyrodnieniem kory mózgowej. [8]
Późne rozpoznanie kiły, przerwanie leczenia, słabe monitorowanie i stany osłabiające kontrolę odporności (niedobory odporności) prowadzą do postępu choroby. [9]
Czynniki ryzyka
- Nieleczona lub niewłaściwie leczona kiła, zwłaszcza w późniejszych stadiach.
- Stany niedoboru odporności.
- Nadużywanie substancji psychoaktywnych, bariery społeczne w dostępie do leków.
- Podeszły wiek, długi okres od zakażenia. [10]
Tabela 3. Kto jest szczególnie narażony
| Kategoria | Dlaczego ryzyko jest wyższe? |
|---|---|
| Osoby z nieleczoną kiłą | Przetrwanie krętków w ośrodkowym układzie nerwowym |
| Pacjenci z niedoborem odporności | Zmniejszona kontrola zakażeń |
| Pacjenci ze skargami poznawczymi/psychiatrycznymi bez wyraźnej przyczyny | Możliwa jest „maska” postępującego paraliżu |
| Osoby starsze z historią kiły | Długi okres utajony |
Patogeneza
Kiła miąższowa układu nerwowego powoduje przewlekłe zapalenie opon mózgowo-rdzeniowych i mózgu, obejmujące płaty czołowe i skroniowe, co wyjaśnia objawy behawioralne i poznawcze. Morfologicznie obserwuje się utratę neuronów, gliozę, demielinizację i nacieki okołonaczyniowe. Z czasem rozwija się rozlany zanik kory mózgowej i zaburzenia przewodzenia, a otępienie postępuje. [11]
Powiązanym późnym zespołem jest zespół więzadła grzbietowego (włókienka grzbietowe, korzenie): ataksja czuciowa, przeszywające bóle, źrenica Argylla-Robertsona. Często jest on związany z postępującym paraliżem lub nakłada się na niego w czasie, ale obraz kliniczny jest inny. [12]
Objawy
Klasyczna triada: zmiany osobowości, pogorszenie funkcji poznawczych i objawy neurologiczne. Często zaczyna się od apatii, rozhamowania, utraty zdolności oceny sytuacji, depresji lub psychozy oraz upośledzenia funkcji wykonawczych i pamięci. Mogą wystąpić drżenie języka i rąk, dyzartria, niezdarność ruchów precyzyjnych, hiperrefleksja i drgawki. Mogą również wystąpić bóle głowy, zaburzenia snu i objawy pseudobulbarne. [13]
Badanie ujawnia mikroskopijne objawy polisomalne: łagodne drżenie, „drżący” charakter pisma, trudności z liczeniem i pisaniem oraz nieprawidłowe odruchy. Zjawiska źreniczne, takie jak źrenica Argylla-Robertsona, są bardziej charakterystyczne dla źrenicy Tabesa niż dla czystego postępującego porażenia. [14]
Tabela 4. Wskazówki kliniczne „za” postępującym porażeniem
| Blok | Przykłady |
|---|---|
| Psychika i poznanie | Nowe rozhamowanie, utrata krytycznej oceny, upośledzenie pamięci, zaburzenia płata czołowego |
| Neurologia | Dyzartria, drżenie, hiperrefleksja, odruchy patologiczne, drgawki |
| Somatoneurologiczne „drobiazgi” | Drżenie języka, niezręczne pisanie, drobne zdolności motoryczne |
| Anamneza | Historia kiły, niepełne leczenie, czynniki społeczne |
Klasyfikacja, formy i etapy
Ze względu na zespół kliniczny późnego kiła układu nerwowego wyróżnia się:
- Postępujący paraliż (postać miąższowa, mózgowa).
- Tabes dorsalis (guzy i korzenie tylne, rdzeniowe).
- Formy mieszane (GPI + tabes). [15]
Ze względu na przebieg postępujący paraliż tradycyjnie dzieli się na: fazę prodromalną (subiektywne zmiany osobowości), fazę zaawansowaną (demencja, objawy neurologiczne) i fazę terminalną (ciężkie upośledzenie funkcji poznawczych, unieruchomienie). Częstość występowania waha się od kilku miesięcy do kilku lat. [16]
Komplikacje i konsekwencje
Bez leczenia rozwija się nieodwracalna demencja, następuje utrata niezależności, wzrasta ryzyko urazów w wyniku napadów padaczkowych i upadków, a także możliwe jest zachłystowe zapalenie płuc, odleżyny i zakażenia współistniejące. Późna kiła układu nerwowego zwiększa śmiertelność, ale dzięki wczesnemu leczeniu można poprawić niektóre funkcje poznawcze i objawy behawioralne. [17]
Kiedy udać się do lekarza
Pilne – w przypadku nowych napadów padaczkowych, ostrego majaczenia, nagłych zmian w zachowaniu, szybko narastających deficytów poznawczych lub nagłych ogniskowych objawów neurologicznych. Wymaga to natychmiastowej diagnostyki, w tym neuroobrazowania i analizy płynu mózgowo-rdzeniowego. Opcjonalne – w przypadku stopniowych zmian osobowości lub zaburzeń pamięci u osoby z kiłą w wywiadzie. [18]
Diagnostyka
Krok 1. Serologia krwi. Przeprowadza się testy niekrętkowe (np. miano VDRL/RPR) i krętkowe (TPPA/FTA-ABS). Pozytywny wynik pary testów wskazuje na przebytą/aktywną kiłę i wymaga oceny ośrodkowego układu nerwowego. [19]
Krok 2. Płyn mózgowo-rdzeniowy. Wykonuje się nakłucie lędźwiowe: cytoza, białko, VDRL w płynie mózgowo-rdzeniowym (wysoka swoistość), odczyn krętkowy w płynie mózgowo-rdzeniowym (czuły, ale mniej swoisty). Rozpoznanie kiły układu nerwowego potwierdza wzrost stężenia białka/komórek i/lub dodatni odczyn niekrętkowy w płynie mózgowo-rdzeniowym na tle objawów klinicznych. Powtarzane nakłucia służą do monitorowania odpowiedzi na leczenie (zwykle po 6–12 miesiącach). [20]
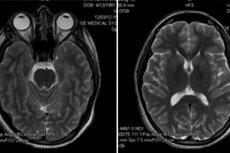
Krok 3. Neuroobrazowanie. Rezonans magnetyczny mózgu może wykazać rozproszony zanik kory mózgowej, zmiany w istocie białej, a czasem wzmocnienie opon mózgowych, ale wyniki są niespecyficzne; są przydatne do wykluczania alternatyw. [21]
Krok 4. Testy neuropsychologiczne. Badania przesiewowe (np. testy stanu psychicznego, testy funkcji wykonawczych) dokumentują stopień deficytów poznawczych i pomagają monitorować poprawę po leczeniu.
Tabela 5. Co uznaje się za potwierdzenie kiły układu nerwowego
| Część | Znaczenie |
|---|---|
| Obraz kliniczny (objawy neurologiczne/psychiczne) | Wymagany |
| Serologia krwi (krętkowe + miano) | Wspomaga diagnozę kiły |
| Płyn mózgowo-rdzeniowy VDRL (dodatni) | Wysoka swoistość dla kiły układu nerwowego |
| Płyn mózgowo-rdzeniowy (cytoza/białko↑, odczyn krętkowy+) | Wsparcie dla negatywnego VDRL |
| Dynamika po leczeniu | Normalizacja płynu mózgowo-rdzeniowego/mian, poprawa kliniczna |
Diagnostyka różnicowa
- Choroba Alzheimera i inne demencje - w przypadku kiły układu nerwowego częściej występuje profil "czołowy" ze zmianami zachowania, napadami padaczkowymi i objawami neurologicznymi.
- Otępienie czołowo-skroniowe ma podobne objawy, ale badania serologiczne w kierunku kiły i badania płynu mózgowo-rdzeniowego stawiają diagnozę.
- Stwardnienie rozsiane, autoimmunologiczne zapalenie mózgu, guzy i wodogłowie normotensyjne można wykluczyć za pomocą MRI i innych specyficznych testów.
- Tabes dorsalis – odmienny profil: ataksja czuciowa, bóle piorunujące, arefleksja, źrenica Argylla Robertsona. [22]
Tabela 6. Postępujący paraliż a więzadło grzbietowe (krótko)
| Podpisać | Postępujący paraliż | Przyssawka grzbietowa |
|---|---|---|
| Dominuje | Zespół poznawczo-psychiczny | Ataksja sensoryczna, bóle przeszywające |
| Uczeń Argylla Robertsona | Rzadko | Często |
| Drgawki | Często | Rzadko |
| Rezonans magnetyczny | Zanik kory mózgowej, niespecyficzny | Zmiany w kręgosłupie, często prawidłowy obraz MRI mózgu |
Leczenie
Leczenie kiły układu nerwowego polega na podaniu penicyliny w dawce wystarczającej do przeniknięcia do płynu mózgowo-rdzeniowego. Zalecany schemat leczenia, zgodnie z wytycznymi Centrów Kontroli i Prewencji Chorób (CDC), to penicylina G rozpuszczalna w wodzie, krystaliczna, podawana dożylnie w dawce 18–24 milionów jednostek na dobę, w dawkach podzielonych co 4 godziny lub w ciągłym wlewie, przez 10–14 dni. Schemat ten jest odpowiedni w przypadku wszystkich postaci kiły układu nerwowego, w tym postępującego porażenia. [23]
Alternatywą (jeśli terapia dożylna nie jest możliwa) może być penicylina prokainowa G w dawce 2,4 mln j.m. domięśniowo raz dziennie + probenecyd 500 mg doustnie 4 razy dziennie przez 10–14 dni, ale ta opcja jest stosowana rzadziej. Po zakończeniu kuracji głównej dopuszczalne jest dodanie penicyliny benzatynowej G w dawce 2,4 mln j.m. domięśniowo raz w tygodniu przez 1–3 tygodnie, tak aby całkowity czas trwania terapii był zbliżony do schematów leczenia kiły późnej. Decyzję podejmuje lekarz. [24]
W przypadku alergii na penicylinę, odczulanie z następowym podaniem penicyliny jest uważane za standardowe leczenie, zwłaszcza u kobiet w ciąży. Dopuszczalne alternatywy (np. ceftriakson) zostały mniej dobrze przebadane; w ich przypadku konieczne jest dokładniejsze monitorowanie odpowiedzi laboratoryjnej i klinicznej. [25]
Na początku leczenia może wystąpić reakcja Jarischa-Herxheimera (gorączka, pogorszenie stanu zdrowia w ciągu pierwszych 24 godzin). Jest to przejściowa reakcja zapalna na obumarcie krętków; zwykle ustępuje samoistnie. W przypadku nasilenia objawów stosuje się leczenie wspomagające. [26]
Ważnym elementem jest monitorowanie po leczeniu. Funkcje poznawcze i behawioralne ocenia się klinicznie, a miana przeciwciał w teście bezkrętkowym (RPR/VDRL we krwi) monitoruje się laboratoryjnie, spodziewając się czterokrotnego spadku w ciągu 6–12 miesięcy. W przypadku niekorzystnej dynamiki lub utrzymujących się dolegliwości, nakłucie lędźwiowe powtarza się (zwykle po 6–12 miesiącach) i rozważa się drugi cykl leczenia. [27]
Terapia objawowa i rehabilitacja obejmują kontrolę napadów padaczkowych za pomocą leków przeciwdrgawkowych, leczenie depresji/psychozy, trening poznawczy oraz współpracę z logopedą i terapeutą zajęciowym. Przy odpowiedniej terapii etiotropowej u niektórych pacjentów w ciągu kilku miesięcy obserwuje się poprawę zachowania, uwagi i pamięci, choć w zaawansowanych przypadkach odwracalność zmian jest ograniczona. [28]
Wsparcie psychiatryczne pomaga radzić sobie z lękiem, agresją, objawami urojeniowymi i afektywnymi oraz poprawia bezpieczeństwo pacjenta i innych osób. Rodziny są edukowane na temat charakteru choroby i przewidywanego przebiegu zdarzeń, co zmniejsza stygmatyzację i zwiększa przestrzeganie zaleceń terapeutycznych. [29]
Jeżeli u pacjenta występują objawy mieszanej późnej postaci (np. tabes + GPI), schemat etiotropowy jest taki sam, a w części objawowej włącza się środki mające na celu kontrolę bólu neuropatycznego i zespołu ataksji (laski, ortezy, ćwiczenia równoważne). [30]
Ciąża i koinfekcje wymagają leczenia wielodyscyplinarnego; penicylina pozostaje lekiem pierwszego wyboru, a odpowiednie leczenie matki zapobiega kile wrodzonej. Nawet jeśli u kobiety w ciąży rozpozna się kiłę układu nerwowego, wczesne leczenie może chronić płód. [31]
Wreszcie, zapobieganie przenoszeniu i ponownemu zakażeniu jest częścią leczenia. Pacjentowi zaleca się badanie partnerów seksualnych, stosowanie antykoncepcji barierowej, monitorowanie w kierunku innych chorób przenoszonych drogą płciową oraz przestrzeganie harmonogramu wizyt kontrolnych. [32]
Tabela 7. Terapia kiły układu nerwowego (podsumowanie dla pacjenta i lekarza)
| Element | Zalecenie |
|---|---|
| Pierwsza linia | Penicylina G rozpuszczalna w wodzie 18-24 mln jednostek/dobę dożylnie przez 10-14 dni |
| Alternatywa (jeśli dożylne podanie leku nie jest dostępne) | Penicylina prokainowa 2,4 miliona jednostek/dzień domięśniowo + probenecyd 500 mg 4 razy dziennie przez 10–14 dni |
| Po kursie (opcjonalnie) | Penicylina benzatynowa G 2,4 miliona jednostek domięśniowo co tydzień przez 1-3 tygodnie |
| Alergia na penicylinę | Odczulanie i penicylina; alternatywy - indywidualnie |
| Monitorowanie | Obraz kliniczny + miana RPR/VDRL; w razie konieczności powtórzenie nakłucia |
Zapobieganie
Skuteczne zapobieganie postępującemu paraliżowi obejmuje wczesne wykrycie i pełne leczenie kiły w jej wczesnym stadium. Obejmuje to regularne badanie osób z grupy ryzyka, edukację, metody barierowe, powiadamianie partnerów oraz ścisłe przestrzeganie odstępów między zastrzykami penicyliny benzatynowej w leczeniu późnej kiły. [33]
W przypadku pacjentów, u których wystąpiła już kiła układu nerwowego, ważne jest regularne wykonywanie badań laboratoryjnych w celu wykrycia w porę braku odpowiedzi serologicznej i przeprowadzenia ponownego leczenia. [34]
Prognoza
Bez leczenia postępujący paraliż prowadzi do ciężkiej demencji i niepełnosprawności. Terapia może zahamować postęp choroby, a także poprawić niektóre zaburzenia poznawcze i behawioralne; stopień odwracalności zależy od czasu trwania procesu i początkowego poziomu uszkodzeń. Wczesna diagnoza i przestrzeganie schematu leczenia penicyliną są kluczem do pomyślnego wyniku. [35]
Tabela 8. Co decyduje o wyniku
| Czynnik | Wpływ na rokowanie |
|---|---|
| Okres od początku do leczenia | Im krócej, tym lepiej się regeneruje |
| Stopień deficytu poznawczego na początku | Związane z mniejszą odwracalnością |
| Przestrzeganie terapii i monitorowanie | Zmniejsza ryzyko nawrotu/postępu choroby |
| Koinfekcje i choroby somatyczne | Pogorszyć rokowanie |
Często zadawane pytania
Czy to choroba „psychiczna”?
Nie. To zakaźne zaburzenie mózgu wywołane przez kiłę. Objawia się objawami psychicznymi i poznawczymi, ale przyczyną jest krętek, a leczenie polega na podawaniu antybiotyków. [36]
Czy możliwe jest całkowite wyleczenie?
Treponema jest znanym powikłaniem, wymagającym odpowiedniej terapii penicyliną. Stopień powrotu funkcji poznawczych zależy od czasu trwania i ciężkości zakażenia. [37]
Czy punkcja płynu mózgowo-rdzeniowego (PMR) jest konieczna?
Tak, jest to standard w potwierdzaniu kiły układu nerwowego i monitorowaniu odpowiedzi na leczenie. [38]
Dlaczego po dożylnych wstrzyknięciach dodaje się „cotygodniowe zastrzyki”?
Aby „dodać” całkowity czas trwania terapii do późnej kiły; opcja ta jest przewidziana w wytycznych i omawiana z lekarzem. [39]
Jakie testy są potrzebne?
Z kim się skontaktować?