Ekspert medyczny artykułu

Nowe publikacje
Wścieklizna u dzieci
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Wścieklizna, zwana też wścieklizną, to ostra choroba wirusowa przenoszona poprzez ugryzienie przez zakażone zwierzę. Powoduje uszkodzenie układu nerwowego i rozwój ciężkiego zapalenia mózgu zakończonego zgonem.
Epidemiologia
Wirus wścieklizny, będący plagą zdrowia publicznego od czasów starożytnych, obecnie powoduje około 59 000 zgonów ludzi rocznie, z których prawie wszystkie są przenoszone przez pogryzienia psów. Ma to znaczący wpływ ekonomiczny na kraje rozwijające się, szczególnie w Afryce i Azji, które mogą ponieść najmniejsze straty. Jednak pomimo prawie 100-procentowego wskaźnika śmiertelności, wścieklizna u psów jest chorobą całkowicie zapobiegawczą, a historyczne przykłady eradykacji wścieklizny u psów w rozwiniętym świecie to potwierdzają. [ 1 ]
Przyczyny wścieklizna
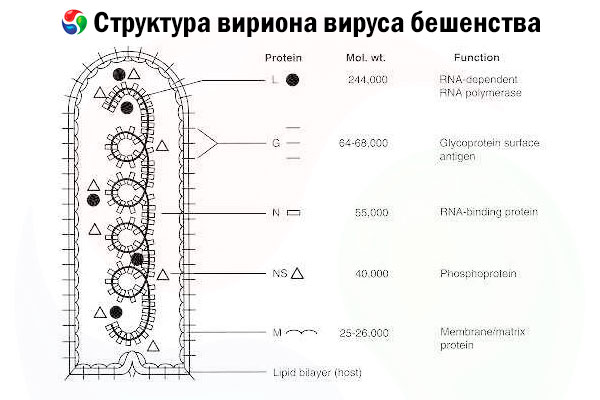
Czynnikiem sprawczym jest wirus wścieklizny (RV), wirus o ujemnej polarności RNA z rodziny rabdowirusów, o rozmiarach około 60 nm × 180 nm.
Składa się z wewnętrznego rdzenia białkowego, czyli nukleokapsydu, zawierającego kwas nukleinowy, oraz zewnętrznej błony, zawierającej lipidy dwuwarstwy pokrytej kolcami glikoproteiny transbłonowej. Ma stosunkowo prostą modułową strukturę genomu i koduje pięć białek strukturalnych:
- Polimeraza RNA zależna od RNA (L),
- nukleoproteina (N)
- fosforylowane białko (P)
- białko macierzy (M) i
- zewnętrzna glikoproteina powierzchniowa (G).
Białka N, P i L wraz z genomowym RNA tworzą kompleks rybonukleoproteinowy. G jest jedynym antygenem RV zdolnym do indukowania produkcji przeciwciał neutralizujących RV, które są głównymi efektorami odpornościowymi przeciwko śmiertelnemu zakażeniu RV. Z drugiej strony wykazano, że kompleks rybonukleoproteinowy jest głównym antygenem RV zdolnym do indukowania komórek CD4+ T, które mogą zwiększać produkcję przeciwciał neutralizujących RV poprzez wewnątrzstrukturalne rozpoznanie antygenu. [ 2 ] Kompleks rybonukleoproteinowy może odgrywać ważną rolę w ustanawianiu pamięci immunologicznej i długoterminowej odporności. [ 3 ]
 [
[ Klasyfikacja i typy antygenów
Rodzaj Lyssavirus obejmuje wirus wścieklizny oraz antygenowo i genetycznie spokrewnione wirusy wścieklizny: wirusy nietoperzy Lagos, Mokola i Duvenhage, a także dwa domniemane podtypy europejskich lyssavirusów nietoperzy. Badania ochrony krzyżowej wskazują, że zwierzęta szczepione tradycyjnymi szczepionkami przeciwko wściekliźnie mogą nie być w pełni chronione, gdy zostaną wystawione na działanie innych lyssavirusów.
Wirusy wścieklizny można klasyfikować jako stałe (zaadaptowane przez pasażowanie u zwierząt lub hodowlę komórkową) lub uliczne (typ dziki). Zastosowanie przeciwciał monoklonalnych i sekwencjonowania genetycznego w celu odróżnienia wirusów ulicznych wścieklizny pomogło w identyfikacji wariantów wirusowych pochodzących z głównych rezerwuarów żywicieli na całym świecie i w sugerowaniu prawdopodobnych źródeł narażenia ludzi, gdy w przypadku pacjenta nie było w innym przypadku historii jednoznacznego ugryzienia przez zwierzę.[ 8 ]
Patogeneza
Głównym rezerwuarem i źródłem zakażenia wśród zwierząt dzikich są wilki, lisy, szakale, nietoperze, a wśród zwierząt domowych - psy i koty, rzadko - konie, bydło, świnie, szczury itp. Przenoszenie zakażenia z człowieka na człowieka, chociaż możliwe, jest niezwykle rzadkie. Jest to typowa infekcja odzwierzęca. Ludzie zarażają się wścieklizną głównie od psów.
Po ugryzieniu człowieka przez chore zwierzę wirus namnaża się w tkance mięśniowej w miejscu ukąszenia, a następnie, po dotarciu do końców nerwów obwodowych czuciowych, rozprzestrzenia się dośrodkowo, docierając do neuronów ruchowych. Czas potrzebny wirusowi na przemieszczenie się i zaatakowanie mózgu zależy od miejsca ukąszenia. W przypadku poważnych ukąszeń głowy i twarzy wirus może dotrzeć do ośrodkowego układu nerwowego w ciągu 15-20 dni, a w przypadku niewielkich uszkodzeń skóry tułowia i kończyn, a w konsekwencji niewielkiej dawki patogenu, proces przemieszczania się wirusa do ośrodkowego układu nerwowego może zostać opóźniony o kilka miesięcy, a nawet do 1-1,5 roku. Po dotarciu do ośrodkowego układu nerwowego wirus utrwala się w tkankach mózgu i rdzenia kręgowego, głównie w neuronach rdzenia przedłużonego, rogu Ammona i podstawy mózgu. W rdzeniu kręgowym najbardziej dotknięte są rogi tylne. Z ośrodkowego układu nerwowego wirus przemieszcza się odśrodkowo wzdłuż pni nerwowych do gruczołów ślinowych, gdzie się namnaża i jest wydalany ze śliną.
Koncepcje patogenezy wścieklizny
RV ma szeroki zakres żywicieli i może zarażać niemal wszystkie ssaki. Chociaż odnotowano kilka dróg transmisji RV, naturalna infekcja najczęściej następuje poprzez ugryzienie. Oprócz ugryzień, spożycie padliny zakażonej RV może sprzyjać zakażeniu wirusem wścieklizny u lisów polarnych, a kontakt RV z błonami śluzowymi okazał się inną możliwą drogą transmisji. [ 9 ] W niektórych nietypowych okolicznościach, takich jak przypadkowe uwolnienie RV w postaci aerozolu w laboratorium lub RV w postaci aerozolu w jaskiniach zamieszkiwanych przez dużą liczbę nietoperzy, [ 10 ] może dojść do transmisji aerozolowej.
Nie jest jeszcze jasne, czy uliczny RV i szczepy RV przystosowane do myszy lub hodowli tkankowej replikują się w miejscu szczepienia przed przedostaniem się do OUN. Podczas gdy eksperymentalne domięśniowe zakażenie młodych chomików lub szopów ulicznym RV wykazało replikację RV w komórkach mięśni prążkowanych zanim wirus zaatakował aksony neuronów ruchowych przez połączenia nerwowo-mięśniowe, [ 11 ], [ 12 ] domięśniowe zakażenie myszy zaadaptowanym do myszy CVS-24 RV wykazało, że RV migruje bezpośrednio do OUN bez wcześniejszej replikacji w miejscu szczepienia. [ 13 ] Po dostaniu się do zakończeń niezmielinizowanych aksonów, RV jest wstecznie transportowany do ciała komórki.
Najnowsze odkrycia sugerują, że transport pęcherzyków aksonowych może stanowić kluczową strategię dla dalekosiężnego przemieszczania się wirionów w aksonach. [ 14 ] Oszacowano, że RV migruje w aksonach z szybkością 3 mm/h. [ 15 ] Następnie infekcja rozprzestrzenia się przez łańcuch neuronów połączonych połączeniami synaptycznymi. Jednak dokładny mechanizm, który promuje transsynaptyczne rozprzestrzenianie się, jest nadal nieznany. Po zakażeniu mózgu wirus rozprzestrzenia się odśrodkowo do obwodowego i autonomicznego układu nerwowego w wielu narządach obwodowych. [ 16 ] W ostatnim etapie cyklu infekcji RV migruje do gruczołów ślinowych; po replikacji w mucogennych komórkach pęcherzykowych jest uwalniany do śliny i jest gotowy do przeniesienia do następnego gospodarza. [ 17 ]
W odniesieniu do patologii wywołanej wirusem wścieklizny, apoptotyczna śmierć komórek została zaproponowana jako potencjalny mechanizm patogenny w eksperymentalnych modelach wścieklizny u myszy zakażonych ustalonym szczepem RV. [ 18 ] Mechanizmem patogennym, który może przyczyniać się do głębokiej dysfunkcji ośrodkowego układu nerwowego charakterystycznej dla wścieklizny, może być upośledzona funkcja neuronalna. Wykazano, że ekspresja genów jest wyraźnie zmniejszona w neuronach zakażonych RV, co skutkuje ogólnym zahamowaniem syntezy białek, [ 19 ] a kilka badań wykazało upośledzoną neurotransmisję po zakażeniu RV. Jiang wykazał, że wiązanie antagonisty receptora acetylocholiny do zakażonych homogenatów mózgu szczura było zmniejszone w porównaniu z grupą kontrolną. [ 20 ] Zaburzone uwalnianie i wiązanie serotoniny, neuroprzekaźnika zaangażowanego w kontrolę cyklu snu, percepcję bólu i zachowanie, obserwowano również w mózgu szczura zakażonego RV. [ 21 ], [ 22 ] Oprócz wpływu na neurotransmisję, zakażenie prawej komory może również wpływać na kanały jonowe. Zakażone komórki mysiego neuroblastoma wykazują zmniejszoną ekspresję funkcjonalną kanałów sodowych bramkowanych napięciem, co może zapobiegać potencjałom czynnościowym i ostatecznie prowadzić do upośledzenia czynnościowego. [ 23 ]
Oprócz braku poważnych zmian patologicznych w ośrodkowym układzie nerwowym, większość przypadków wścieklizny u ludzi nie wywołuje odpowiedzi immunologicznej 7 do 10 dni po wystąpieniu objawów klinicznych. Te głębokie różnice między patogenezą wścieklizny a patogenezą większości innych wirusowych lub bakteryjnych infekcji ośrodkowego układu nerwowego są dodatkowo poparte faktem, że immunosupresja jest nieskuteczna lub szkodliwa dla wyniku wścieklizny. [ 24 ] Niski poziom odpowiedzi immunologicznej często obserwowany u ofiar wścieklizny jest zagadkowy, ponieważ nie można go wyjaśnić słabą immunogennością antygenów RV. W rzeczywistości RV G i białko nukleokapsydu są silnymi antygenami komórek B i T po podaniu pozajelitowym. [ 25 ] Możliwym wyjaśnieniem niskiego stopnia odpowiedzi immunologicznej przeciwko RV u ludzi i zwierząt chorych na wściekliznę może być to, że zakażenie RV ośrodkowego układu nerwowego powoduje immunosupresję, [ 26 ] i zaproponowano, że RV stosuje strategię wywrotową, obejmującą zapobieganie apoptozie i niszczenie atakujących komórek T. [ 27 ]
Atenuowane szczepy RV, które zostały przystosowane do komórek nieneuronalnych, różnią się znacząco od patogennych szczepów ulicznych RV pod względem neuroinwazyjności, która odnosi się do ich zdolności do inwazji OUN z miejsc obwodowych. Pod tym względem szczepy RV przystosowane do hodowli tkankowych albo nie mają, albo mają tylko ograniczoną zdolność do inwazji OUN z miejsc obwodowych, podczas gdy szczepy uliczne RV lub szczepy RV przystosowane do myszy, takie jak CVS-24, są wysoce inwazyjne. [ 28 ] Kluczowe czynniki zaangażowane w neuroinwazję RV obejmują wychwyt wirusa, transport aksonalny, rozprzestrzenianie się transsynaptyczne i szybkość replikacji wirusa.
Do niedawna nasza wiedza na temat patogenezy RV była ograniczona i opierała się głównie na badaniach opisowych ulicznych szczepów RV lub eksperymentalnych zakażeniach atenuowanymi szczepami zaadaptowanymi w laboratorium. Pojawienie się technologii odwrotnej genetyki pozwoliło nam zidentyfikować elementy wirusowe, które determinują patogenny fenotyp RV i lepiej zrozumieć mechanizmy zaangażowane w patogenezę wścieklizny.
Identyfikacja elementów wirusowych kontrolujących nabywanie, rozprzestrzenianie i replikację wirusa wścieklizny
- Elementy wirusowe biorące udział w wychwytywaniu wirusów
Zakażenie RV rozpoczyna się od przyłączenia wirusa do domniemanego receptora komórkowego. Chociaż zaproponowano kilka cząsteczek powierzchni błony jako receptory RV, w tym receptor nikotynowy acetylocholiny[ 29 ], cząsteczkę adhezji komórek nerwowych[ 30 ] i receptor neurotrofinowy o niskim powinowactwie p75NTR[ 31 ], nadal nie jest jasne, czy te cząsteczki faktycznie odgrywają rolę w cyklu życia wirusa wścieklizny. W tym kontekście niedawno wykazano, że interakcja RV G–p75NTR nie jest wymagana do zakażenia pierwotnych neuronów RV.[ 32 ] Po związaniu receptora RV jest internalizowany poprzez endocytozę adsorpcyjną lub zależną od receptora. [ 33 ] Środowisko o niskim pH w przedziale endosomalnym wywołuje następnie zmiany konformacyjne w RV G, które wyzwalają fuzję błony wirusowej z błoną endosomalną, uwalniając w ten sposób RNP do cytoplazmy. [ 34 ] W przypadku wirusów RV G odgrywa kluczową rolę w wychwycie wirusa, najprawdopodobniej poprzez interakcje z domniemanymi receptorami komórkowymi, które ułatwiają szybkie wychwytywanie. W tym względzie wykazano, że patogenność szczepów RV przystosowanych do hodowli tkankowych (np. ERA, HEP i CVS-11) koreluje z obecnością determinanty zlokalizowanej w miejscu antygenowym III białka G. [ 35 ] Mutacja Arg → Gln w pozycji 333 w tym miejscu antygenowym białka ERA G spowodowała siedmiokrotne opóźnienie internalizacji wariantu Gln333 RV w porównaniu z wariantem typu dzikiego. Mutacja Asn194→Lys194 w RV G, która wyjaśnia ponowne pojawienie się fenotypu patogennego, była związana ze znacznym skróceniem czasu internalizacji.[ 36 ] Co więcej, eksperymenty z chimerycznymi RV wykazały, że czas potrzebny do internalizacji wirionów RV znacznie się wydłużył, a patogenność znacznie spadła po zastąpieniu genu G wysoce patogennego szczepu SB RV, który został uzyskany z klonu cDNA szczepu RV-18 pochodzącego ze srebra, związanego z nietoperzami,[ 37 ] genem wysoce atenuowanego szczepu SN, który został wyizolowany z klonu cDNA szczepu szczepionkowego SAD B19 RV.[ 38 ] Łącznie dane te wspierają pogląd, że kinetyka pobierania wirusa, która jest funkcją RV G, jest głównym czynnikiem determinującym patogenność RV.
- Elementy wirusowe biorące udział w rozprzestrzenianiu się i transmisji wirusów
Unikalną właściwością wirusa wścieklizny jest jego zdolność do rozprzestrzeniania się z komórki do komórki. Obserwacja, że wariant ERA Gln333 traci zależną od pH aktywność fuzji komórka-komórka in vitro [ 39 ] i wykazuje znacznie zmniejszoną zdolność do rozprzestrzeniania się z komórki do komórki [ 40 ] sugeruje, że RV G odgrywa również kluczową rolę w rozprzestrzenianiu się z komórki do komórki, a zatem w transmisji wirusa, prawdopodobnie poprzez swoją aktywność fuzjogenną. Możliwość tę dodatkowo potwierdza odkrycie, że szybkość rozprzestrzeniania się patogennego rewertanta RV SPBNGAK jest prawie dwukrotnie wyższa niż ta ustalona dla niepatogennego wariantu SPBNGA. Co ciekawe, mutacja Asn 194 → Lys 194 w G SPBNGAK spowodowała przesunięcie progu pH dla fuzji błon na wyższe pH, co potwierdza hipotezę, że wyższy próg pH dla fuzji błon jest związany ze zwiększonym rozprzestrzenianiem się wirusa. [ 41 ]
Badania transneuronalnych wskaźników zakażenia RV u szczurów [ 42 ] i makaków rezus [ 43 ] wykazały, że wirus wścieklizny migruje wyłącznie w kierunku wstecznym w aksonach. Chociaż kilka białek RV bierze udział w mechanizmach transportu neuronalnego, RV G wydaje się odgrywać dominującą rolę w transneuronalnym rozprzestrzenianiu się zakażenia RV. Na przykład, podczas gdy obwodowe zakażenie wirusem zakaźnej niedokrwistości koni (EIAV) pseudotypowanym RV G skutkuje przeniesieniem wirusa do rdzenia kręgowego, ten sam EIAV pseudotypowany wirusem zapalenia pęcherzykowego G nie przedostał się do układu nerwowego. [ 44 ] Ponadto stwierdzono, że rozprzestrzenianie się wirusa mutanta ERA G Arg 333 → Gln 333 w OUN jest znacznie zmniejszone w porównaniu z mutantem dzikiego typu, co dodatkowo sugeruje funkcję nienaruszonego RV G w rozprzestrzenianiu transsynaptycznym. Jednakże najbardziej przekonujący dowód na ważną rolę RV G w transporcie transsynaptycznym pochodzi z wewnątrzczaszkowej infekcji myszy rekombinowanym wirusem RV z niedoborem G, który wykazał, że infekcja pozostała ograniczona do neuronów w miejscu szczepienia bez żadnych dowodów na rozprzestrzenienie się na neurony wtórne. [ 45 ] Jednakże prawdopodobne jest, że oprócz RV G, RV M odgrywa również rolę w rozprzestrzenianiu się wirusa, a zatem w transporcie transsynaptycznym. W tym względzie wykazano, że rozprzestrzenianie się chimerycznego wariantu SN-BMBG RV, który zawiera zarówno M, jak i G z wysoce patogennego SB, było znacznie wyższe niż rozprzestrzenianie się chimerycznego wariantu SN-BG lub SN-BM, które zawierają odpowiednio G i M z SB, co sugeruje, że optymalna interakcja M z G może odgrywać ważną rolę w rozprzestrzenianiu się wirusa z komórki do komórki. [ 46 ] Ponieważ RV M wspomaga pączkowanie wirusa, [ 47 ] prawdopodobne jest, że bardziej wydajne rozprzestrzenianie się chimerycznej odmiany RV SN-BMBG wynika z optymalnego pączkowania wirusa na błonie postsynaptycznej.
Najnowsze badania wykazały, że interakcja między RV P a łańcuchem lekkim dyneiny łączy RV RNP z układem transportu komórkowego gospodarza, ułatwiając w ten sposób wsteczny transport aksonalny wirusa.[ 48 ],[ 49 ] Jednakże obwodowe zakażenie dorosłych myszy wykazało, że usunięcie domeny wiążącej LC8 RV P nie zapobiega przedostaniu się wirusa do ośrodkowego układu nerwowego, co sugeruje, że białko RV nie bierze bezpośredniego udziału w wstecznym rozprzestrzenianiu się aksonalnym RV.[ 50 ]
- Elementy wirusowe kontrolujące replikację wirusa
W przeciwieństwie do wielu innych wirusów, takich jak wirus grypy, patogeniczność RV jest odwrotnie proporcjonalna do szybkości syntezy wirusowego RNA i produkcji zakaźnych cząstek wirusowych. Porównanie poziomów wirusowego mRNA i genomowego RNA wytwarzanych przez różne wirusy chimeryczne sugeruje, że transkrypcja i replikacja wirusowego RNA są regulowane przez wiele czynników, w tym RV M, który został zidentyfikowany jako czynnik trans-działający, który pośredniczy w przejściu z początkowo wysokich poziomów syntezy mRNA do syntezy genomowego RNA. [ 51 ] Ponadto, M ze wszystkich rabdowirusów jest w stanie wyłączyć ekspresję genów wirusowych poprzez wiązanie się z RNP, co skutkuje utworzeniem wysoce skondensowanej struktury przypominającej szkielet, która nie jest w stanie wspierać syntezy RNA.
Aby zidentyfikować inne elementy wirusowe, które kontrolują patogeniczność poprzez regulację replikacji wirusa, sekwencje końcowe 5' wysoce patogennego szczepu SB były stopniowo zastępowane sekwencjami z wysoce atenuowanego szczepu szczepionkowego SN, co skutkowało rekombinowanymi wirusami SB2 (sekwencja końcowa [TS] + L), SB3 (TS + L + pseudogen [Ψ]), SB4 (TS + L + Ψ + G) i SB5 (TS + L + Ψ + G + M). Zakażenie domięśniowe rodzicielskimi wirusami SB i SN oraz chimerycznymi RV SB2, SB3, SB4 i SB5 wywołało najwyższe wskaźniki śmiertelności u myszy zakażonych SB i brak zachorowań lub śmiertelności u myszy zakażonych SN. Zastąpienie TS, L i SB odpowiednimi elementami z SN skutkowało umiarkowaną redukcją zachorowań i śmiertelności, a dodatkowa wymiana G lub G plus M silnie zmniejszyła lub całkowicie zniosła patogeniczność wirusa.
Fenotypowa charakterystyka tych dzikich i chimerycznych RV w hodowli tkankowej wykazała, że patogeniczność danego RV jest odwrotnie skorelowana z jego zdolnością do replikacji w komórkach neuronalnych. Chociaż SB replikował się na poziomach prawie 1000-krotnie niższych niż SN, a zastąpienie TS, L i w SB poziomami SN miało niewielki wpływ na kinetykę wzrostu wirusa, dodatkowe zastąpienie G lub G plus M w SB przez odpowiadające im geny SN spowodowało wzrost produkcji wirusa o 1 log, co sugeruje, że kinetyka replikacji wirusowego RNA, jak również produkcja cząstek wirusowych są w dużej mierze kontrolowane przez białko G RV. Wniosek ten jest poparty danymi uzyskanymi z wariantami RV G, które różnią się jednym aminokwasem w swoich białkach G. Patogenny wariant wirusa wścieklizny SPBNGAK 194 wytworzył miano wirusa w komórkach NA, które było o 1 log niższe niż to wytworzone przez niepatogenny wariant SPBNGAN 194, a analiza PCR w czasie rzeczywistym wykazała, że szybkość transkrypcji i replikacji wirusowego RNA w komórkach NA zakażonych SPBNGAK była 5- i 10-krotnie wyższa niż w komórkach NA zakażonych SPBNGAK. [ 52 ] Dalsze dowody na odwrotną korelację między patogennością a szybkością syntezy wirusowego RNA i produkcji cząstek wirusowych dostarczyły myszy zakażone chimerycznymi wirusami rekombinowanymi, w których geny G i M atenuowanego szczepu SN zostały zastąpione genami wysoce patogennego szczepu SB. Eksperymenty te ujawniły znaczący wzrost patogenności rodzicielskiego szczepu SN przenoszącego RV G w porównaniu do patogennego szczepu SB. Patogeniczność wzrosła jeszcze bardziej, gdy do SN wprowadzono zarówno G, jak i M z SB.
Podstawienie G lub M lub obu w SN odpowiednimi genami z SB wiązało się ze znacznym spadkiem tempa produkcji cząstek wirusowych, jak również tempa syntezy wirusowego RNA. Dane te wskazują, że zarówno G, jak i M odgrywają ważną rolę w patogenezie RV, regulując replikację wirusa. Odkrycie, że podstawienie G lub G plus M w SN przez G lub G plus M z SB skutkuje umiarkowanym do silnego spadkiem transkrypcji i replikacji wirusowego RNA, odpowiednio, podczas gdy podstawienie samego M w SN przez M z SB skutkuje silnym wzrostem transkrypcji i replikacji wirusowego RNA, wskazuje, że RV G ma również ważną funkcję regulacyjną w transkrypcji/replikacji wirusowego RNA, samodzielnie lub poprzez interakcję z białkiem M. Mechanizm, za pomocą którego gen RV G kontroluje syntezę wirusowego RNA, jest nieznany. Pewne sekwencje nukleotydowe w genach RV G, takie jak te obejmujące kodony dla Arg333 i Lys 194, zostały zidentyfikowane jako cele dla komórkowych miRNA. Wykazano, że rozpoznanie celu przez miRNA komórkowe może skutkować pozytywną lub negatywną regulacją replikacji wirusa. [ 53 ] Podstawienia Arg 333 → Glu 333 lub Lys 194 → Ser 194 w sekwencji genu G RV skutkują zniesieniem sekwencji docelowych miRNA, co z kolei wiąże się ze znacznym wzrostem szybkości syntezy wirusowego RNA [Faber M, Thomas Jefferson University, PA, USA, niepublikowane dane], co sugeruje, że miRNA komórkowe gospodarza odgrywają również ważną rolę w regulacji replikacji RV, co wykazano w przypadku innych wirusów RNA, w tym wirusa zapalenia pęcherzykowego jamy ustnej i HCV. [ 54 ], [ 55 ]
Regulacja replikacji wirusa wydaje się być jednym z ważnych mechanizmów zaangażowanych w patogenezę RV. Aby uniknąć odpowiedzi immunologicznej i zachować integralność sieci neuronowej, patogenne szczepy RV, ale nie szczepy atenuowane, mogą regulować tempo swojego wzrostu. Niższe tempo replikacji prawdopodobnie przynosi korzyści patogennym szczepom RV poprzez zachowanie struktury neuronowej, której te wirusy używają do dotarcia do OUN. Innym wyjaśnieniem niższego tempa replikacji patogennego RV jest to, że w celu uniknięcia wczesnego wykrycia przez układ odpornościowy gospodarza wirus utrzymuje minimalne poziomy ekspresji swoich antygenów.
Związek między ekspresją RV G, apoptozą i patogenicznością
Dobrze wiadomo, że szczepy wirusa wścieklizny ulicznej, które są znacznie bardziej patogenne niż szczepy przystosowane do hodowli tkankowej, wyrażają bardzo ograniczone poziomy G i nie indukują apoptozy aż do późnego etapu cyklu zakaźnego, co sugeruje, że patogenność konkretnego szczepu wirusa jest odwrotnie skorelowana z ekspresją RV G i zdolnością do indukowania apoptozy. [ 56 ] Bezpośredni dowód na korelację między poziomem ekspresji G i stopniem apoptozy uzyskano w przypadku rekombinowanego RV SPBNGA-GA, który przenosił dwa identyczne geny G i nadmiernie ekspresjonował RV G. [ 57 ] Badania morfologiczne kultur neuronalnych zakażonych tym rekombinowanym RV wykazały, że śmierć komórek była znacząco zwiększona równolegle z nadmierną ekspresją RV G i że apoptoza jest głównym mechanizmem zaangażowanym w śmierć pośredniczoną przez RV G. W szczególności zmniejszenie barwienia F-aktyną po zakażeniu SPBNGA-GA jest zgodne z depolimeryzacją włókien aktynowych indukowaną apoptozą. Co więcej, liczba jąder TUNEL-pozytywnych w neuronach zakażonych SPBNGA-GA była znacząco zwiększona w porównaniu do neuronów niezakażonych i zakażonych SPBNGA. Jednak mechanizm, za pomocą którego gen RV G pośredniczy w procesie sygnalizacji apoptozy, pozostaje w dużej mierze nieznany. Zasugerowano, że ekspresja RV G powyżej pewnego progu poważnie zakłóca błonę komórkową. Jest wysoce prawdopodobne, że komórki apoptotyczne nie są szybko usuwane z OUN i dlatego ulegają wtórnej martwicy. [ 58 ] Z drugiej strony, zakażenie RV, a w szczególności nadmierna ekspresja białka RV G, może prowadzić do piroptozy, ścieżki śmierci komórki podobnej do apoptozy, która w przeciwieństwie do apoptozy obejmuje aktywację kaspazy 1 i tym samym prowadzi do martwicy. [ 59 ] Stopień martwicy lub piroptozy wywołanej zakażeniem RV prawdopodobnie odgrywa kluczową rolę w indukcji odporności przeciwwirusowej. Podczas gdy komórki apoptotyczne zachowują integralność błony komórkowej i nie stymulują wrodzonej odpowiedzi immunologicznej, komórki martwicze stają się przepuszczalne i wydzielają endogenne adiuwanty, które mogą wywołać silną wrodzoną odpowiedź immunologiczną. [ 60 ]
Ponieważ poziom apoptozy/martwicy koreluje z immunogennością RV, zasugerowano, że immunostymulujący wpływ komórek apoptotycznych/martwiczych najprawdopodobniej przyczynia się do generowania ochronnej odpowiedzi immunologicznej. Dlatego regulacja ekspresji RV G jest bardzo prawdopodobnie ważnym czynnikiem w patogenezie wścieklizny, ponieważ zapewnia sposób na przeżycie i rozprzestrzenianie się patogennych wariantów RV w układzie nerwowym bez powodowania jawnego uszkodzenia neuronów i wywoływania ochronnej odpowiedzi immunologicznej, która zapobiegłaby zakażeniu.
Ekspresja RV G może być regulowana na poziomie syntezy RNA, poziomie potranslacyjnym lub obu. Wykazano, że poziomy RV G wyrażane przez różne warianty chimeryczne RV są odzwierciedlone przez szybkość syntezy wirusowego RNA, co sugeruje, że różnicowa regulacja ekspresji RV G przez te warianty wynika ze zmian w szybkości transkrypcji wirusowego mRNA. Podobnie jak w przypadku szybkości transkrypcji wirusowego RNA, ilość RV G wyrażana przez te warianty odwrotnie koreluje z patogennością wirusa. Z drugiej strony, zakażenie pierwotnych kultur neuronalnych mniej patogennym wariantem RV CVS-B2c skutkowało czterokrotnie wyższymi poziomami białka G niż zakażenie wysoce patogennym wariantem CVS-N2c, pomimo syntezy porównywalnych poziomów mRNA G w obu zakażeniach. Eksperymenty typu „pulse-chase” wykazały, że wyższe poziomy białka G w neuronach zakażonych CVS-B2c były w dużej mierze wynikiem niższej szybkości degradacji białka G CVS-B2c w porównaniu z białkiem G CVS-N2c. Mechanizm prowadzący do szybszej proteolitycznej degradacji białka G CVS-N2c pozostaje jednak niewyjaśniony.
Objawy wścieklizna
Okres inkubacji wścieklizny wynosi średnio 30-90 dni. W przypadku masywnego zakażenia przez duże rany głowy i twarzy może on zostać skrócony do 12 dni. W rzadkich przypadkach okres inkubacji może trwać 1 rok lub dłużej.
Następuje ściśle sekwencyjna zmiana trzech okresów choroby: prodromalnego, pobudzenia, porażenia.
Okres prodromalny rozpoczyna się pojawieniem bólu bolącego lub ciągnącego w miejscu ukąszenia, a także bólu wzdłuż nerwów. W okolicy blizny może wystąpić uczucie pieczenia, swędzenia, czasami zaczerwienienia i obrzęku. Pacjent odczuwa ogólne złe samopoczucie, ból głowy, nudności. Obserwuje się wymioty, wzrost temperatury ciała do 37,5-38 °C oraz objawy postępującego zaburzenia psychicznego: wzmożoną pobudliwość odruchową, niewytłumaczalne uczucie lęku, strachu, melancholii. Często pacjent jest przygnębiony, zahamowany, wycofany, odmawia jedzenia, źle śpi, skarży się na ponure myśli, przerażające sny. Okres prodromalny trwa 2-3 dni, czasami przedłuża się do 7 dni. Pod koniec tego okresu mogą wystąpić ataki lęku z krótkotrwałymi trudnościami w oddychaniu, uczuciem ucisku w klatce piersiowej, któremu towarzyszy tachykardia i zwiększona częstość oddechów.
Okres podniecenia charakteryzuje się pojawieniem się wścieklizny: przy próbie picia, a następnie na widok wody lub jej przypomnienie, pacjent doświadcza drgawkowego skurczu gardła i krtani, podczas którego z krzykiem wyrzuca kubek z wodą, rzuca do przodu drżące ręce, odrzuca głowę i ciało do tyłu. Szyja jest wyciągnięta, bolesny grymas zniekształca twarz, która staje się sina z powodu skurczu mięśni oddechowych. Oczy wychodzą na wierzch, wyrażają strach, proszą o pomoc, źrenice są rozszerzone, wdychanie jest utrudnione. W szczytowym momencie ataku możliwe jest zatrzymanie akcji serca i oddychania. Atak trwa kilka sekund, po czym stan pacjenta wydaje się poprawiać. Następnie ataki skurczów mięśni krtani i gardła mogą wystąpić nawet od ruchu powietrza (aerofobia), jasnego światła (światłowstręt) lub głośnego słowa (akustykofobia). Atakom towarzyszy pobudzenie psychoruchowe, podczas którego pacjent zachowuje się jak „szaleniec”. Świadomość jest zamglona podczas ataku, ale ustępuje w okresie międzynapadowym. W okresie pobudzenia, ze względu na wzmożone napięcie układu współczulnego, pacjenci doświadczają gwałtownego wzrostu wydzielania śliny (sialorrhea) z niemożnością połykania śliny z powodu skurczu mięśni gardła. Pacjent pryska śliną. U niektórych pacjentów mogą wystąpić objawy meningizmu, a nawet opistotonus, a drgawki są częste. W tym przypadku płyn mózgowo-rdzeniowy może się nie zmieniać, ale u niektórych pacjentów może wzrosnąć stężenie białka i liczba komórek może wzrosnąć z powodu limfocytów.
Bez odpowiedniego leczenia objawy odwodnienia nasilają się, rysy twarzy stają się ostrzejsze, a masa ciała spada. Temperatura ciała wzrasta do wysokich wartości. Możliwe są drgawki. Czas trwania fazy pobudzenia wynosi około 2-3 dni, rzadko 4-5 dni. Zazwyczaj w trakcie jednego z ataków następuje zgon. Rzadko pacjent przeżywa do trzeciego stadium choroby.
W okresie paraliżu pacjent uspokaja się. Napady wścieklizny ustępują, pacjent może pić i połykać pokarmy, świadomość jest jasna. Jednak pomimo pozornego dobrego samopoczucia, nasilają się letarg, apatia, depresja, wkrótce pojawiają się porażenia kończyn, zaburzenia miednicy, porażenie nerwów czaszkowych. Temperatura ciała wzrasta do 42-43 °C, spada ciśnienie tętnicze, a pod koniec pierwszego dnia następuje śmierć z powodu porażenia ośrodków sercowo-naczyniowego i oddechowego.
We krwi obwodowej obserwuje się leukocytozę neutrofilową, zwiększoną liczbę hemoglobiny, erytrocytów i hematokrytu.
Co Cię dręczy?
Formularze
Klinicznie rozróżnia się formy typowe i nietypowe. Formy nietypowe obejmują wszystkie przypadki bez pobudzenia i hydrofobii. Formy nietypowe obejmują bulbar, cerebellar, meningoencephalitic, itp.
Diagnostyka wścieklizna
Wykrycie antygenu wścieklizny, przeciwciał, RNA wirusa lub izolacji wirusa umożliwia diagnozę wścieklizny. Ponieważ każdy indywidualny test może być ujemny u pacjenta z wścieklizną, czasami konieczne są seryjne próbki surowicy w celu wykrycia przeciwciał wścieklizny, próbki śliny w celu hodowli wirusa oraz biopsja skóry w celu przeprowadzenia bezpośredniego testu immunofluorescencyjnego w celu wykrycia antygenu wirusa, zwłaszcza gdy istnieje duże podejrzenie wścieklizny.
Jedną z najszybszych metod diagnozowania wścieklizny u ludzi przed śmiercią jest wykonanie bezpośredniego testu immunofluorescencji na biopsji skóry z karku w celu wykrycia antygenu wścieklizny. Bezpośredni test immunofluorescencji jest najczulszą i najspecyficzną metodą wykrywania antygenu wścieklizny w skórze i innych świeżych tkankach (np. biopsja mózgu), chociaż wyniki mogą być czasami ujemne na wczesnym etapie choroby. Jeśli świeża tkanka jest niedostępna, trawienie enzymatyczne utrwalonych tkanek może zwiększyć reaktywność testu immunofluorescencji; jednak czułość może być niedopuszczalnie niska.
Diagnozę można również ustalić, jeśli wirus zostanie wyizolowany ze śliny po zaszczepieniu komórek neuroblastoma lub gryzoni laboratoryjnych; jest to zazwyczaj najskuteczniejsze w ciągu pierwszych 2–3 tygodni choroby. Wykrycie przeciwciał neutralizujących wirusa wścieklizny, zwykle wykonywane za pomocą szybkiego testu hamowania ognisk fluorescencyjnych (RFFIT), w surowicy osób niezaszczepionych, jest również diagnostyczne. Obecność przeciwciał w płynie mózgowo-rdzeniowym potwierdza diagnozę, ale mogą one pojawić się 2–3 dni później niż przeciwciała surowicy i dlatego mogą być mniej przydatne we wczesnych stadiach choroby. Podczas gdy odpowiedź serologiczna po szczepieniu jest na ogół nieodróżnialna od odpowiedzi serologicznej wywołanej przez chorobę, szczepienie zwykle nie powoduje wytwarzania przeciwciał przeciwko płynowi mózgowo-rdzeniowemu.
Tylko siedem przypadków „wyzdrowienia” z wścieklizny w ciągu ostatnich 25 lat zostało dobrze udokumentowanych. Chociaż wirusa wścieklizny nie wyizolowano u żadnego z pacjentów, wysokie miana przeciwciał neutralizujących wściekliznę w próbkach surowicy i obecność przeciwciał neutralizujących w płynie mózgowo-rdzeniowym zdecydowanie potwierdziły diagnozę.
Co trzeba zbadać?
Jakie testy są potrzebne?
Diagnostyka różnicowa
Diagnozę wścieklizny u ludzi zazwyczaj stawia się na podstawie danych epidemiologicznych i klinicznych, a następnie potwierdza się ją w laboratorium. Diagnoza jest prosta, jeśli w wywiadzie występują ukąszenia zwierząt i wystąpiło pełne spektrum objawów i oznak. W przeciwnym razie konieczna jest staranna, ale szybka ocena cech epidemiologicznych i klinicznych mniej typowych przypadków przed wykonaniem konkretnych badań laboratoryjnych. Każdego pacjenta z objawami neurologicznymi lub niewyjaśnionym zapaleniem mózgu należy zapytać o możliwość narażenia na kontakt ze zwierzętami na obszarach endemicznych dla wścieklizny w kraju zamieszkania lub poza nim. Brak podejrzenia wścieklizny w przypadku kilku niedawnych zgonów ludzi w Stanach Zjednoczonych mógł wynikać z braku starannej historii narażenia.
Na początku choroby wścieklizna może imitować wiele chorób zakaźnych i niezakaźnych. Wiele innych zapaleń mózgu, takich jak te wywoływane przez wirusy opryszczki i arbowirusy, przypomina wściekliznę. Inne choroby zakaźne mogą również imitować wściekliznę, takie jak tężec, malaria mózgowa, riketsjoza i dur brzuszny. Paralityczne choroby zakaźne, które można pomylić ze wścieklizną, obejmują polio, botulizm i zapalenie mózgu wywołane przez małpy opryszczkowe typu B.
Choroby niezakaźne, które mogą być mylone ze wścieklizną, obejmują szereg zespołów neurologicznych, zwłaszcza ostrą polineuropatię zapalną (zespół Guillaina-Barrégo), a także alergiczne zapalenie mózgu i rdzenia po szczepieniu, wtórne do szczepienia tkanki nerwowej przeciwko wściekliźnie, zatrucie lub zatrucie lekami, odstawienie alkoholu, ostrą porfirię i histerię wścieklizny. Zespół Guillaina-Barrégo może być mylony z porażenną wścieklizną i odwrotnie.
Z kim się skontaktować?
Leczenie wścieklizna
Leczenie wścieklizny nie zostało opracowane. Podawanie dużych dawek swoistej immunoglobuliny przeciw wściekliźnie i interferonu leukocytarnego jest nieskuteczne. Leczenie objawowe stosuje się w celu złagodzenia cierpienia pacjenta. W tym celu pacjenta umieszcza się w oddzielnej sali lub boksie, tworzy się reżim ochronny, który ogranicza wpływ środowiska zewnętrznego (zmniejszony hałas, jasne światło, przepływ powietrza). Aby zmniejszyć pobudliwość ośrodkowego układu nerwowego, przepisuje się tabletki nasenne, leki przeciwdrgawkowe i przeciwbólowe. Normalizuje się gospodarkę wodną.
W fazie porażennej przepisuje się leki pobudzające aktywność układu sercowo-naczyniowego i oddechowego. Zaleca się stosowanie hiperbarii tlenowej, hipotermii mózgowej, kontrolowanego oddychania mechanicznego z całkowitą kuraryzacją pacjenta. Jednak wszystkie metody leczenia są praktycznie nieskuteczne. W najlepszym przypadku możliwe jest przedłużenie życia pacjenta o kilka miesięcy. Niekorzystny wynik jest z góry przesądzony ciężkością uszkodzenia pnia mózgu z zniszczeniem ośrodków witalnych.
Zapobieganie
Opracowanie pierwszej szczepionki przeciwko wściekliźnie przez Pasteura w 1885 r. zapoczątkowało erę znacznie skuteczniejszej kontroli wścieklizny. Obecnie, pomimo prawie 100-procentowego wskaźnika śmiertelności u ludzi z powodu wścieklizny, chorobie można całkowicie zapobiec poprzez szczepienie przed i/lub po narażeniu. Podczas gdy Pasteur i jego współpracownicy zainicjowali szczepienie prywatnych psów w Paryżu, pierwsze masowe szczepienie psów przeprowadzono na początku lat 20. XX wieku w Japonii, co oznaczało pierwszy duży krajowy program kontroli wścieklizny. Doustne szczepienie dzikich zwierząt, opracowane po raz pierwszy w latach 70. XX wieku, od tego czasu wielokrotnie wykazano, że skutecznie kontroluje chorobę u głównych żywicieli lądowych, takich jak lisy, szopy i skunksy. [ 68 ] Utrzymujące się szczepienie przeciwko wściekliźnie populacji zwierząt rezerwuarowych przy wskaźnikach pokrycia 70% lub wyższych ostatecznie wyeliminuje RABV z gatunków rezerwuarowych i zapobiegnie rozprzestrzenianiu się wirusa na przypadkowych żywicieli. [ 69 ]
Dane filogenetyczne wskazują, że lyssavirusy zarażały nietoperze na długo przed zakażeniem ssaków lądowych, a większość lyssavirusów, w tym RABV, nadal krąży u różnych gatunków nietoperzy na całym świecie. [ 70 ] Jednak skuteczne metody zapobiegania przenoszeniu RABV wśród nietoperzy pozostają nieuchwytne, co wyklucza możliwość całkowitej eradykacji wścieklizny w tym momencie. Jednak nawet po narażeniu na RABV poprzez ugryzienie ssaka zakażonego wścieklizną, bezpieczna i skuteczna profilaktyka poekspozycyjna (PEP, w tym czyszczenie ran, immunoglobulina przeciw wściekliźnie i szczepienie przeciw wściekliźnie) może chronić ludzi przed zakażeniem wścieklizną, jeśli leczenie zostanie podane szybko i zgodnie z zaleceniami Światowej Organizacji Zdrowia (WHO).
Te dwie metody zapobiegania śmierci ludzi — jedna oparta na szczepieniu narażonych osób, a druga oparta na szczepieniu wystarczającej liczby psów, aby przerwać cykl transmisji u źródła — są podstawowymi elementami podejścia „jednego zdrowia” do zapobiegania i kontroli wścieklizny u psów. Te dwa różne sposoby zapobiegania śmierci ludzi były rozważane jako oddzielne alternatywy: Strategia A, oparta na zapewnianiu PEP ludziom i Strategia B, oparta na szczepieniu psów; lub jako składniki połączonej Strategii A + B w analizie prawdopodobnych kosztów alternatywnych strategii.[ 71 ]
Kraje takie jak Tajlandia odniosły ogromny sukces w zapobieganiu śmierci ludzi dzięki stosowaniu PEP, ale również zauważyły wzrost zapotrzebowania i związanych z tym kosztów związanych ze stosowaniem samej PEP. [ 72 ] Na przykład w porównaniu z sytuacją w 1991 r. w 2003 r. PEP potrzebowało cztery razy więcej osób (ponad 400 000). Najnowsze dane pokazują, że Chińska Republika Ludowa, która szczepi 15 milionów osób rocznie po potencjalnym narażeniu na wściekliznę, wydaje około 650 milionów dolarów amerykańskich rocznie na samą PEP. [ 73 ]
Znacznie bardziej zrównoważonym podejściem jest zapobieganie rozprzestrzenianiu się infekcji u źródła, w populacji zwierząt, przy jednoczesnym zwiększaniu dostępu do PEP dla narażonych pacjentów ludzkich, gdy jest to konieczne. Tam, gdzie istnieje wola polityczna i odpowiednie finansowanie w celu kontrolowania wścieklizny u psów, śmiertelność może zostać wyeliminowana i została wyeliminowana. Powszechne stosowanie szczepień psów doprowadziło do wyeliminowania wścieklizny u psów w kilku krajach, w tym w Malezji w 1954 r., [ 74 ] Japonii w 1956 r., na Tajwanie w 1961 r., w Singapurze, a w szczególności w całej Europie Zachodniej (przegląd w Rupprecht i in., King i in. oraz Gongal i Wright). [ 75 ]
Использованная литература