
Syndrom Pierre Robin
Ostatnia recenzja: 23.04.2024

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.

Syndrom Pierre Robin, znany również w medycynie jako anomalia Robina, jest wrodzoną patologią rozwoju części szczęki twarzy. Jego nazwa została odebrana na cześć francuskiego dentysty P. Robina, który pierwszy opisał wszystkie jego znaki. Lannelongue i Menard po raz pierwszy opisali syndrom Pierre'a Robina w 1891 r. W jego raporcie na przykładzie 2 pacjentów z mikrognatą, rozszczepem podniebienia i retroglossoptozą. W 1926 roku Pierre-Robin opublikował przypadek choroby u dziecka z objawami klasycznego zespołu. Do 1974 r. Triada objawów była znana jako zespół Robin-Pierre'a. Niemniej jednak, ten zespół jest obecnie używany do opisu defektów formacji, podczas gdy występują liczne anomalie.
Epidemiologia
Jest to heterogenna wada wrodzona, której rozpowszechnienie wynosi 1 na 8.500 żywych urodzeń. Stosunek mężczyzn do kobiet wynosi 1: 1, z wyjątkiem formy chromosomu X.
Wśród tych pacjentów, u 50% niemowląt rozszczep podniebienia miękkiego jest niepełny, reszta rodzi się z łukowatym i niezwykle wysokim niebem, ale bez rozszczepu.
Przyczyny syndrom Pierre Robin
Rozważa się możliwość autosomalnego recesywnego dziedziczenia choroby. Istnieją dwa typy syndromów, w zależności od etiologii: izolowane i uwarunkowane genetycznie. Pojedyncze gatunki rozwijają się z powodu uciskania dolnej części szczęki podczas rozwoju embrionalnego. Kompresja może rozwijać się z powodu:
- Obecność w macicy lokalnych pieczęci (cyst, blizn, guzów).
- Ciąża mnoga.
Również rozwój żuchwy u płodu może być osłabiony, gdy:
- Infekcje wirusowe, które przyszła matka cierpiała w czasie ciąży.
- Zaburzenia neurotroficzne.
- Niewystarczające ilości kwasu foliowego w ciele kobiety w ciąży.
Patogeneza
Zespół Pierre Robin objawia się z powodu zaburzeń embrionalnych, które są spowodowane różnymi patologiami w okresie prenatalnym.
Istnieją trzy teorie patofizjologiczne, które mogą wyjaśnić pojawienie się syndromu Pierre'a Robina.
Teoria mechaniki : ta teoria jest najbardziej prawdopodobna. Niedorozwój aparatu żuchwy występuje między 7 a 11 tygodniem ciąży. Wysoka pozycja języka w jamie ustnej prowadzi do powstawania rozpadlin na niebie, z tego powodu nie ma zamykania płyt podniebiennych. Ta teoria wyjaśnia klasyczną odwróconą szczelinę w kształcie litery U i związaną z nią wargę zająca. W etiologii rola może odgrywać rolę małowodzie, ponieważ brak płynu owodniowego może prowadzić do odkształcenia podbródka i następującego po nim ucisku języka między płytkami podniebiennymi.
Teoria neurologiczna : Opóźnienie w rozwoju neurologicznym odnotowano w elektromiografii języka języka i gardłowych kolumnach oraz smaku z powodu opóźnienia przewodzenia w nerwie gnykowym.
Teoria regulacji Disneya w kształcie rombu : Teoria ta opiera się na naruszeniu rozwoju romboidalnego mózgu w procesie ontogenezy.
Niedostateczny rozwój dolnej części szczęki dziecka prowadzi do znacznego zmniejszenia jamy ustnej. To z kolei powoduje tak zwaną pseudomakroglossię, czyli język jest przemieszczany w kierunku tylnej części ściany gardła. Ta patologia prowadzi do rozwoju niedrożności dróg oddechowych.
Dopóki dziecko płacze lub się porusza, drożność dróg oddechowych pozostaje normalna, ale gdy zasypia, znowu pojawia się przeszkoda.
Z powodu zaburzeń oddechowych proces karmienia dziecka jest bardzo trudny. W tej chwili prawie zawsze występuje niedrożność dróg oddechowych. Jeśli nie zastosujesz korekty medycznej, taka patologia może doprowadzić do poważnego wyczerpania całego organizmu, a nawet do zgonu.
Objawy syndrom Pierre Robin
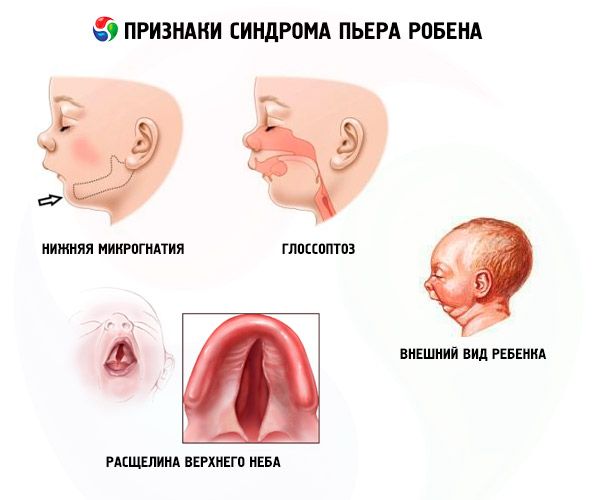
Choroba charakteryzuje się trzema głównymi objawami:
- Niższa mikrognatologia (niewystarczający rozwój żuchwy występuje w 91,7% przypadków). Charakteryzuje się cofnięciem dolnego łuku zębowego o 10-12 mm za górnym łukiem. Żuchwa ma małe ciało, kąt rozwarty. Dziecko osiąga normalny rozwój w wieku około 5-6 lat.
- Glossoptosis (utrata języka z powodu jej niewystarczającego rozwoju odnotowano w 70-85% przypadków).
- Makroglossia i ankyloglossia są względnie rzadkimi objawami, odnotowanymi w 10-15% przypadków.
- Na niebie pojawia się szczelina.
- Bradypnea i duszność.
- Jasna sinica.
- Asfiksja, która najczęściej objawia się podczas prób karmienia dziecka.
- Połknięcie jest niemożliwe lub bardzo trudne.
- Pragnienie wymiotów.
- Anomalie uszne w 75% przypadków.
- Utraty słuchu charakteru stwierdzono u 60% pacjentów, a zarośnięcie zewnętrznego przewodu słuchowego znajduje się tylko 5% pacjentów, brak Pneumatyzacja z sutkowatego jamie kości w czasie.
- Anomalie ucha wewnętrznego (aplazja bocznych kanałów półkolistych, duży akwedukt przedsionkowy, utrata komórek włosowych ślimaka).
- Wady nosa są rzadkie i są głównie reprezentowane przez anomalie korzenia nosa.
- Wady rozwojowe zębów występują w 30% przypadków. Niedoczynność krtani i neo-gardła obserwuje się u około 10-15% pacjentów z zespołem Pierre'a Robina.
Systemowe objawy zespołu Pierre Robin
Systemowe nieprawidłowości rozwojowe opisano w 10-85% zgłoszonych przypadków.
Anomalie oka występują u 10-30% pacjentów. Mogą występować: nadwzroczność, krótkowzroczność, astygmatyzm, stwardnienie rogówki i zwężenie kanału nosowo-łzowego.
Patologie sercowo-naczyniowe: łagodny szmer sercowy, zwężenie tętnicy płucnej, otwarty przewód tętniczy, owalne okno, ubytek przegrody międzyprzedsionkowej i nadciśnienie płucne. Ich rozpowszechnienie waha się od 5-58%.
Anomalie związane z zaburzeniami ruchu (70-80% przypadków): syndaktylii, dysplazji paliczka, polidaktylia, klinodaktylia, nadmiernej ruchomości stawów i oligodaktiliya górnych. Anomalie kończyn dolnych: stopy anomalie (Club stopy, przodostopia wskazywaniu), deformacja kości udowej (koślawego lub Varus biodra, krótkie kości udowej), zaburzenia wrodzone zwichnięcie stawu biodrowego (przykurcze), z nieprawidłowości kolanowych (GENU koślawego, chrząstkozrost). Wady rozwojowe kręgosłupa: skolioza, kifoza, lordoza, dysplazja kręgów, agenezji sacrum i kości ogonowej zatokowego.
Patologia ośrodkowego układu nerwowego: epilepsja, opóźniony rozwój układu nerwowego, wodogłowie. Częstotliwość defektów OUN wynosi około 50%.
Nieprawidłowości w układzie moczowo-płciowym: nie upadłe jądra (25%), wodonercze (15%), a także rozlane jądra (10%).
Powiązanych zespołów i warunki: zespół sticklera, zespół trisomią 11q, trisomia 18, zespół delecji 4q, reumatoidalne artropatia, gipohondroplaziya, zespół Moebiusa.
Gradacja
Istnieją trzy etapy nasilenia choroby, które zależą od stanu dróg oddechowych dziecka:
- Łatwo - są małe problemy z karmieniem, ale oddychanie nie jest prawie trudne. Leczenie odbywa się w warunkach ambulatoryjnych.
- Średni - oddech jest umiarkowanie trudny, karmienie dziecka jest umiarkowanie trudne. Zabieg przeprowadza się w szpitalu.
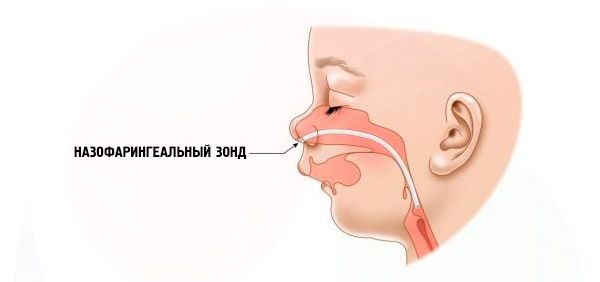
- Ciężki oddech jest bardzo trudny, dziecko nie może być karmione normalnie. Konieczne jest stosowanie specjalnych urządzeń (sonda donosowa).
Komplikacje i konsekwencje
Połączenie mikrognathy i glossoptosis może prowadzić do poważnych powikłań oddechowych i problemów podczas karmienia dziecka.
Zespół Pierre Robin powoduje następujące komplikacje:
- Streezing oddychanie z powodu niedrożności dróg oddechowych. Laryngomalacja, a nawet asfiksja we śnie.
- Rozwój psychoruchowy dziecka pozostaje daleko w tyle za rówieśnikami.
- Rozwój fizyczny również pozostaje w tyle.
- Mowa u pacjentów jest zepsuta.
- Częste choroby zakaźne w uchu, które stają się przewlekłe i prowadzą do uszkodzenia słuchu.
- Zespół obturacyjnego bezdechu sennego, początek śmierci podczas snu zmienia się w 14-91% przypadków.
- Problemy z zębami.
Diagnostyka syndrom Pierre Robin
Rozpoznanie zespołu Pierre Robin nie powoduje trudności. Opiera się na objawach klinicznych. Aby wykluczyć inne patologie, bardzo ważne jest skonsultowanie się z genetykiem.
Dzieci z wrodzoną anomalią Robin od urodzenia odpadł z powodu ciągłej stagnacji języka. Dziecko zachowuje się niespokojnie, skóra jest sinoczuła, a wdech z klatki piersiowej. Podczas karmienia może wystąpić uduszenie. Diagnoza może być również przeprowadzona zgodnie z nietypowym wyglądem dziecka - "twarzą ptaka". Często inni pacjenci mają inne wady: krótkowzroczność, zaćmę, patologię układu moczowo-płciowego, patologię serca, anomalie w rozwoju kręgosłupa.
W przypadku tych objawów klinicznych nietrudno postawić właściwą diagnozę specjalistom.
Z kim się skontaktować?
Leczenie syndrom Pierre Robin
Leczenie odbywa się natychmiast po urodzeniu dziecka z zespołem Pierre Robin. Jeśli choroba jest łagodna, to aby poprawić stan pacjenta, należy stale trzymać dziecko w pozycji pionowej lub leżeć na brzuchu. Głowa dziecka powinna być przechylona do klatki piersiowej. Podczas karmienia nie zaleca się trzymania dziecka w pozycji poziomej, aby pokarm nie dostał się do dróg oddechowych.
Jeśli niewystarczająca ekspresja dolnej części szczęki jest wyrażona dość mocno, interwencja chirurgiczna jest stosowana w celu doprowadzenia tonącego języka do normalnej fizjologicznej pozycji. W ciężkich przypadkach język jest zaciśnięty i przymocowany do dolnej wargi. W bardzo ciężkich przypadkach konieczne jest wykonanie tracheostomii, połyskopeksja, dystrakcyjna osteogeneza żuchwy.
Stosowane jest również leczenie zachowawcze.
Leki
Fenobarbital. Śpiący i uspokajający lek, różni działanie przeciwdrgawkowe. W każdej tabletce jest 100 ml fenobarbitalu. Dawkowanie jest indywidualne, ponieważ zależy od ciężkości choroby i stanu dziecka. Pacjenci z niewydolnością wątroby, hiperkinezją, niedokrwistością, miastenią rzekomą, porfirią, cukrzycą, depresją, nietolerancją składników leku są zabronione. Przy podejmowaniu następujących objawów są możliwe: zawroty głowy, osłabienie, halucynacje, agranulocytoza, nudności, niskie ciśnienie krwi, alergie.
Clonazepam. Lek przepisany w leczeniu padaczki. Lek zawiera substancję czynną klonazepam będący pochodną benzodiazepiny. Różni się działanie przeciwdrgawkowe, przeciwlękowe i miorelaksiruyuschim. Dawka jest ustalana przez lekarza prowadzącego, ale nie powinna przekraczać maksymalnej dawki - 250 mcg na dobę. Nie należy przyjmować bezsenności, hypertonii mięśniowej, pobudzenia psychoruchowego, lęku napadowego. Przy podejmowaniu następujących objawów są możliwe: opóźnienie, nudności, bolesne miesiączkowanie, ból głowy, leukopenia, opóźnienie lub nietrzymanie moczu, łysienie, alergia.
Sibazon. Wyprodukowany w postaci roztworu i tabletek doodbytniczych. Substancją czynną jest pochodna benzodiazepiny (sibazon). Różni się działaniem uspokajającym, przeciwlękowym, przeciwdrgawkowym. Dawkowanie jest indywidualne. Pacjenci z przewlekłą hiperkapnią, myasthenia gravis, nietolerancja na benzodiazepiny nie powinni przyjmować leku. Podczas stosowania leku możliwe jest wystąpienie takich objawów: nudności, zaparcia, ból głowy, zawroty głowy, czkawka, nietrzymanie moczu, alergie.
Cortexin jest liofilizatem. Lek o działaniu nootropowym. Lek zawiera kompleks frakcji polipeptydowych rozpuszczalnych w wodzie i glicyny. Dawkowanie jest indywidualne i jest przepisywane przez lekarza prowadzącego w zależności od stanu pacjenta. Pacjenci z nietolerancją koryksyny nie powinni przyjmować tego leku. Produkt może powodować reakcje alergiczne.
Leczenie fizjoterapeutyczne
Co do zasady, w stadium stadium syndromu, terapia pozycyjna jest wykonywana, gdy dziecko jest umieszczane na brzuchu w pozycji pionowej, dopóki siła grawitacji nie spowoduje prawidłowego wzrostu dolnej części szczęki.
Leczenie operacyjne
Leczenie operacyjne stosuje się przede wszystkim do korekcji glossoptosis. Istnieje kilka metod:
- Wsparcie ze srebrną nicią języka. Nić jest przenoszona przez dolną część dziąsła i dolną wargę. Metoda nazywa się Douglas.
- Metoda Duhamela - gruby srebrny nić przenosi się przez podstawę języka pacjenta i dwa policzki. Użyj nie dłużej niż trzydzieści dni.
- Urządzenia ortopedyczne do rysowania i mocowania języka.
- W wieku jednego roku można wykonać operację eliminacji rozszczepienia na niebie.
Prognoza
Prognozy i przebieg choroby są poważne. Najczęściej w pierwszych dniach życia, ze średnią i ciężką fazą choroby, następuje śmierć (przyczyną jest uduszenie). Również ryzyko śmiertelnego wyniku w pierwszym roku jest dość wysokie z powodu licznych infekcji.
U pacjentów w wieku po dwóch latach prognoza jest korzystna.
 [36]
[36]