
Zespół Tholosa-Hunta
Ostatnia recenzja: 18.10.2021

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
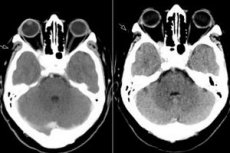
Syndrom szczeliny oczodołu górnego, patologiczna oftalmoplegia - to nic innego jak zespół Tholosa Hunta, czyli uszkodzenie struktur w szczelinie oczodołu górnego. Proces ten obejmuje zwykle naczynia oczodołowe (tętnicze i żylne), włókna nerwowe (nerwy okoruchowe, bloczkowe, odwodzące, pierwsza gałąź nerwu trójdzielnego) oraz pobliską zatokę jamistą. Chorobę można zaliczyć do stosunkowo rzadkich i dość trudnych w diagnostyce patologii. [1]
Epidemiologia
Zespół Tholosa Hunta został opisany nie tak dawno: około 70 lat temu. Dokonał tego hiszpański lekarz neurologii E. Tolos. Kilka lat później pracę uzupełnił Anglik, doktor okulistyki W. Hunt. Nazwiska lekarzy badaczy stały się podstawą nazwy zespołu.
Zespół Tholosa Hunta występuje z równą częstością zarówno u mężczyzn, jak iu kobiet. Patologia jest zwykle jednostronna i jest równie często zauważana po lewej lub prawej stronie. Zespół obustronny jest możliwy, ale występuje tylko w pojedynczych przypadkach.
Średni wiek chorych to 50 lat. Ogólnie zespół Tholosa Hunta można zarejestrować w wieku 15-85 lat. Większość pacjentów należy do kategorii wiekowej osób starszych: rozwój choroby sprzyjają licznym zaburzeniom sercowo-naczyniowym, a także związanym z wiekiem zmianom w tkankach.
Najczęstszym objawem choroby jest przejaw klasycznego napadu migreny: osoba ma nagły, pulsujący ból głowy z jednej strony, „strzelający” lub „skręcający”, z napromieniowaniem na orbitę. Ponieważ brak jest typowej specyficznej symptomatologii zespołu Tholosa Hunta, patologia jest często nazywana „neurologicznym kameleonem”: diagnoza jest złożona i wymaga odróżnienia od wielu innych chorób.
Pacjenci z zespołem Tholosa Hunta są okresowo spotykani w różnych krajach świata, bez żadnych cech terytorialnych ani sezonowych. Współczynnik zapadalności wynosi 0,3-1,5 przypadków na 1 milion mieszkańców. [2]
Przyczyny zespół Tholosa-Hunta
W trakcie identyfikacji przyczyn rozwoju zespołu Tholosa Hunta naukowcy odkryli następujące fakty:
- w większości przypadków choroba była wywoływana przez immunologiczne zapalenie zewnętrznej ściany zatoki jamistej;
- w niektórych przypadkach przyczyną były malformacje naczyniowe, procesy nowotworowe w mózgu (formy pierwotne i wtórne), miejscowe zapalenie pachymenergii czaszki, zapalenie mięśni oczodołu, guzkowe zapalenie tętnic i tworzenie się skrzepliny w zatoce jamistej;
- U około 30% pacjentów nie jest możliwe ustalenie przyczyny zaburzenia, dlatego postawiono rozpoznanie idiopatycznego zespołu Tholosa Hunta.
Rozważmy bardziej szczegółowo te rzekome przyczyny.
- Rozwój autoimmunologiczny zespołu jest związany zarówno z hipotermią, jak i niedawnymi patologiami zakaźnymi, a także z głębokimi stresami. Postać autoimmunologiczną choroby charakteryzuje się: ostrym początkiem, nawrotowym przebiegiem, wysoką skutecznością terapii glikokortykosteroidami. Ta postać choroby występuje częściej u mężczyzn.
- W przypadku zdekompensowanego nadciśnienia tętniczego często występują malformacje naczyniowe. Częściej chorują kobiety. Choroba zaczyna się ostro, ból jest umiarkowany, praktycznie bez objawów wytrzeszczu i chemozy.
- Wśród procesów nowotworowych, które mogą prowadzić do rozwoju zespołu Tholosa Hunta, częściej występowały pierwotne guzy mózgu, guzy przerzutowe z pierwotnymi ogniskami w płucach, oskrzelach, prostacie lub przerzuty czerniaka skóry.
- Zlokalizowane pachymenitis czaszki powoduje ostry początek zespołu przy braku objawów mózgowych i oponowych, bez wytrzeszczu. Rozpoznanie potwierdza morfologicznie biopsja.
- Zapalenie mięśni oczodołu powoduje podostry początek, z intensywnym bólem i wytrzeszczem, wyraźną chemozą i podwójnym widzeniem.
- Zakrzepica zatoki jamistej wywołuje rozwój całkowitej oftalmoplegii. Rozpoznanie potwierdza rezonans magnetyczny.
- Guzkowe zapalenie tętnic może spowodować rozwój zespołu Tholosa Hunta kilka miesięcy po wystąpieniu choroby.
Mechanizm autoimmunologiczny w większości przypadków leży u podstaw powstawania patologii, co zostało udowodnione przez wielu ekspertów. W szczególności następujące czynniki wskazują na charakter autoimmunologiczny:
- cykliczny kurs;
- zaburzenia dysimmunologiczne;
- dysocjacja białek w płynie mózgowo-rdzeniowym i zwiększona zawartość cytokin prozapalnych w płynie mózgowo-rdzeniowym i surowicy. [3]
Czynniki ryzyka
Naukowcy nie ustalili jeszcze dokładnej przyczyny pojawienia się zespołu Tholosa Hunta. Ale udało im się zidentyfikować pewne czynniki wpływające na rozwój takiego naruszenia:
- Ogólne predyspozycje genetyczne do chorób autoimmunologicznych. Jeśli jeden z członków rodziny jest chory lub ma chorobę autoimmunologiczną, inni krewni mogą mieć podobne lub inne patologie o podobnym mechanizmie rozwoju. Czynnik ten jest nadal założeniem, które wymaga dodatkowych badań i dowodów.
- Czynniki środowiskowe, w tym nawyki żywieniowe, warunki środowiskowe, jakość wody, zagrożenia przemysłowe itp.
- Ciężkie stresujące sytuacje, częste stresy i wstrząsy psycho-emocjonalne, silne zmiany hormonalne (w tym ciąża, menopauza itp.).
- Długotrwałe przewlekłe choroby zakaźne, w tym zapalenie wątroby, zakażenie wirusem opryszczki, wirus cytomegalii itp.
- Hipotermia, promieniowanie, inne silnie drażniące i szkodliwe czynniki.
Patogeneza
Mechanizm etiologiczny rozwoju zespołu Tholosa Hunta nie został do końca poznany. Decydujące znaczenie przypisuje się reakcjom autoimmunologicznym. Wielu naukowców zakłada, że infekcje wirusowe i bakteryjne, sytuacje stresowe, promieniowanie działają tylko jako czynnik prowokujący. Nie ma mocnych dowodów na związek między spożyciem patogenów a rozwojem zespołu Tholosa Hunta. Istnieją jednak podejrzenia co do udziału wirusa cytomegalii w procesie autoimmunologicznym, który przyczynia się do powstawania ziarniniaków. [4]
Schemat patogenetyczny wynika z pojawienia się miejscowego ziarniniakowego procesu zapalnego w strefie zewnętrznej ściany zatoki jamistej, infraklinoidalnej lub supraklinoidalnej części tętnicy szyjnej wewnętrznej, co prowadzi do jej zwężenia. Ważną rolę odgrywa zaburzenie humoralnej i komórkowej obrony immunologicznej. Humoralna strona zespołu jest związana ze zwiększonym tworzeniem się antyneutrofilnych cytoplazmatycznych przeciwciał, które działają przeciwko enzymom proteinazie-3, mieloperoksydazie i specyficznemu białku błonowemu zdolnemu do wiązania endotoksyn. Przypuszczalnie przeciwciała cytoplazmatyczne stymulują istniejące neutrofile, w wyniku czego atakują docelowe narządy, w szczególności proces zapalny rozwija się w zewnętrznej ścianie zatoki jamistej.
Zmiany komórkowe również odgrywają rolę w rozwoju zespołu Tholosa Hunta. Świadczy o tym dominacja limfocytów T, makrofagów i komórek plazmatycznych w ziarniniakach.
Istnieją informacje o wysoce aktywnych strukturach śródbłonka i cytokinach przeciwzapalnych, co wskazuje na tendencję do przewlekłego przebiegu procesu chorobowego.
W pojedynczych przypadkach stwierdzono ogniskowe zmiany martwicze w obrębie zewnętrznej ściany zatoki jamistej.
Objawy zespół Tholosa-Hunta
Symptomatologia charakterystyczna dla zespołu Tholosa Hunta pojawia się nagle i nieoczekiwanie u samego pacjenta. Główne objawy to:
- Silny ból w okolicy oczodołów, wyjątkowo nieprzyjemny, nudny, rozprzestrzeniający się od okolicy czołowej do łuków brwiowych, oczu i dalej na całą głowę.
- Podwojenie w oczach, które występuje po wystąpieniu bólu. Wizualna koncentracja i rozważenie przedmiotu staje się niezwykle trudne.
- Zaburzenie funkcji motorycznej gałki ocznej, czyli tak zwana oftalmoplegia, głównie jednostronna. Może objawiać się w różnym stopniu, który zależy od ciężkości procesu patologicznego i rozległości zmiany.
- Obrzęk spojówek.
- Przemieszczenie gałki ocznej do przodu (wytrzeszcz, oczy „wyłupiaste”).
- Odchylenie osi wzrokowej jednej gałki ocznej w bok, zez, charakterystyczne dla jednostronnego uszkodzenia nerwu.
- Ogólne pogorszenie stanu zdrowia, niewielki wzrost temperatury, osłabienie, drażliwość.
Obraz kliniczny postępuje stopniowo, objawy zmieniają się i pogarszają, ale mogą zniknąć równie nagle, jak się pojawiły. Jednak w przypadku braku niezbędnej terapii zespół Tholosa Hunta ponownie przypomina o sobie nawrotem.
Objawy neurologiczne wynikają z lokalizacji procesu chorobowego. Ból pojawia się w wyniku podrażnienia pierwszej gałęzi nerwu trójdzielnego, przechodząc w pobliżu pnia nerwu okoruchowego i jest widoczny w oczodole, czole, skroni, podstawie nosa. Intensywność bólu jest różna: od umiarkowanej do ciężkiej.
Możliwe nietypowe objawy, które charakteryzują się brakiem bólu. Można to zaobserwować, gdy zmiana jest zlokalizowana przed wejściem piątej pary do zatoki jamistej.
Zaburzenia okulomotoryczne zwykle objawiają się podwójnym widzeniem podczas bezpośredniego patrzenia.
Jeśli bolesny proces jest zlokalizowany w strefie wierzchołka orbity, często objawy neurologiczne występują w połączeniu z zaburzeniami analizatora wizualnego. W rezultacie pojawia się obrzęk lub zanik nerwu wzrokowego i często obserwuje się mroczki centralne. Możliwy wytrzeszcz (wybrzuszenie), chemoza (obrzęk spojówek), której wystąpienie jest spowodowane naciekowymi zmianami w tkance pozagałkowej i trudnościami z odpływem żylnym z oczodołu.
Pierwsze znaki
Ponieważ zespół Tolosa Hunta nie został dotychczas wystarczająco zbadany, naukowcy nadal wyjaśniają możliwe mechanizmy rozwoju tej patologii. Uwzględniając kryteria nakreślone przez Międzynarodowe Towarzystwo Neurologiczne, rozpoznanie zespołu Tholosa Hunta jest uzasadnione w obecności ziarniniaków zewnętrznej ściany zatoki jamistej, wykrytych podczas MRI mózgu lub biopsji.
Lista objawów, które są akceptowane jako kryteria diagnostyczne zespołu, jest następująca:
- Ból „skaczący” lub „skręcający” w jednym oczodole z następczym rozwojem porażenia mięśni (oftalmoplegia);
- połączone uszkodzenia nerwów okoruchowych, pierwszej gałęzi nerwu trójdzielnego i splotu nerwu okołotętniczego;
- wzrost obrazu klinicznego przez kilka dni (lub w ciągu 1-2 tygodni);
- możliwość spontanicznej remisji (w niektórych przypadkach - z resztkowym zachowaniem wad);
- prawdopodobieństwo ponownego zaostrzenia zespołu po miesiącach lub latach;
- niezmieniony obraz systemowy, brak zmian chorobowych poza zatoką szyjną;
- obecność pozytywnego efektu terapii kortykosteroidami.
Istnieje inna podobna lista diagnostyczna znaków zaproponowana w 2003 roku. Zgodnie z tą listą zespół Tholosa Hunta jest uważany za wynik wzrostu tkanki ziarniniakowej w zatoce jamistej, szczelinie oczodołowej górnej i jamie oczodołowej:
- jeden lub więcej epizodów jednostronnego bolesnego ataku w okolicy oczodołu, które mijają bez leczenia przez kilka tygodni;
- uszkodzenie nerwu czaszkowego (III, IV lub VI) w postaci niedowładów, obecność ziarniniaków, potwierdzone rezonansem magnetycznym lub biopsją;
- pojawienie się niedowładu jednocześnie z zespołem bólowym lub w ciągu 14 dni po nim;
- ustąpienie niedowładu i zespołu bólowego w ciągu 3 dni od rozpoczęcia leczenia kortykosteroidami.
Formularze
W przypadku zespołu Tolosa Hunta lewa i prawa strona są dotknięte mniej więcej równą częstotliwością, dlatego patologia jest podzielona na lewostronną lub prawostronną.
Choroba jest w większości jednostronna. Obustronne uszkodzenie odnotowano tylko w niezwykle rzadkich przypadkach.
Obraz kliniczny choroby może rozwijać się w następujących etapach:
- ostry lub podostry, który występuje po niedawnej wirusowej chorobie zakaźnej, hipotermii, silnym wzroście ciśnienia krwi, czasami bez wyraźnego powodu;
- przewlekłe nawracające, ze stopniowym nasilaniem się objawów i okresowymi zaostrzeniami.
Ponadto zespół Tholosa Hunta może być:
- całkowity, z uszkodzeniem wszystkich nerwów przechodzących przez górną szczelinę oczodołową;
- niekompletne, z zaangażowaniem w patologiczny proces nerwów par VI, IV, III i I gałęzi pary V w różnych kombinacjach.
W odniesieniu do zatoki można wyróżnić przednią, środkową i tylną postać zespołu Tholosa Hunta.
Komplikacje i konsekwencje
Zespół Tholosa Hunta występuje z silnym bólem, który pociąga za sobą utratę snu, zaburzenia w sferze emocjonalnej i psychicznej. Chorzy ludzie stają się rozdrażnieni, niestabilni emocjonalnie. Jeśli niezbędne leczenie nie zostanie przeprowadzone, wówczas na tym tle możliwe jest pojawienie się zaburzeń nerwicowych: rozwijają się stany depresyjne, neurastenia i hipochondria. Zdolność do pracy znacznie spada, pacjent zostaje wycofany.
Cechą charakterystyczną zespołu Tholosa Hunta jest nawracający przebieg, który często występuje w chorobach autoimmunologicznych. Czas trwania remisji jest bardzo zróżnicowany: maksymalny zarejestrowany bezobjawowy czas trwania wynosił 11 lat. Po zabiegu ryzyko nawrotu jest znacznie zmniejszone. Jeśli wystąpią zaostrzenia, różnią się one mniej ciężkim przebiegiem.
Diagnostyka zespół Tholosa-Hunta
Lekarzom często trudno jest natychmiast zdiagnozować zespół Tholosa Hunta, ponieważ objawy są bardzo podobne do innych, bardziej powszechnych chorób. W większości przypadków wymagana jest dodatkowa konsultacja kilku wąskich specjalistów: neuropatologa, okulisty, endokrynologa, onkologa, neurochirurga itp.
W pierwszym etapie należy wykluczyć choroby złośliwe, tętniaki, zapalenie opon mózgowych itp.
Najczęściej zespół Tholosa Hunta określa się metodą wykluczenia: pacjentowi podaje się szereg testów, które wykluczają inne najbardziej prawdopodobne choroby. Wymagane są następujące testy:
- szczegółowy obraz krwi;
- badanie funkcji hormonalnej tarczycy;
- badanie poziomu białka całkowitego we krwi (w celu oceny jakości metabolizmu białek);
- analiza płynu mózgowo-rdzeniowego.
- Diagnostyka instrumentalna polega na wykonaniu takich procedur diagnostycznych:
- obrazowanie metodą rezonansu magnetycznego mózgu i regionu oczodołu, z kontrastem i bez;
- angiografia rezonansu magnetycznego;
- cyfrowa angiografia subtrakcyjna (angiografia dożylna);
- komputerowa tomografia mózgu i oczodołu z kontrastem i bez.
MRI ze wzmocnieniem gadolinu jest metodą z wyboru do oceny THS i może wykazać nieprawidłowy wzrost i wzmocnienie CS przechodzącego przez szczelinę oczodołową górną do wierzchołka oczodołu. Zgłaszane wyniki MRI ważone T1 i T2 są skrajnie zmienne i niespecyficzne. MRI odgrywa kluczową rolę w diagnostyce i pomaga wykluczyć inne powszechne zmiany związane z CS, unikając konieczności wykonywania zabiegów inwazyjnych wysokiego ryzyka, takich jak biopsja SC, jedyny sposób na uzyskanie histopatologicznego potwierdzenia tej choroby. [5]
Badania te pomagają zidentyfikować ślady procesów zapalnych w zatoce jamistej, szczelinie oczodołu górnego lub wierzchołku oczodołu. Ślady zapalenia w okolicy oczodołu na obrazach przekrojowych przy braku porażenia nerwu czaszkowego są uważane za bardziej łagodne z punktu widzenia rokowania.
Niektórym pacjentom z podejrzeniem zespołu Tholosa Hunta zaleca się wykonanie biopsji w celu wykluczenia procesów onkologicznych.
Diagnostyka różnicowa
Praktyka kliniczna wskazuje, że podobne objawy mogą występować w wielu patologiach somatycznych i neurologicznych:
- z mikrobiologicznymi, wirusowymi i grzybiczymi procesami zapalnymi, które obejmują opony mózgowe lub zewnętrzną ścianę zatoki jamistej;
- z procesami nowotworowymi w mózgu i orbicie - na przykład z gruczolakiem przysadki, czaszkogardlakiem, nerwiakiem, oponiakiem skrzydła kości klinowej, z przerzutami do mózgu lub oczodołu;
- z wadami naczyniowymi - w szczególności z tętniakami żylno-tętniczymi, przetokami szyjno-jamistymi itp., a także z rozwarstwieniem gałęzi tętnicy szyjnej wewnętrznej;
- z zakrzepicą, torbielowatymi formacjami zatoki jamistej, chłoniakiem;
- z sarkoidozą, zapaleniem mięśni oczodołu (mięśnie oka), ziarniniakowatością Wegenera (ziarniniakowatością z zapaleniem naczyń), oftalmigreną, niektórymi patologiami krwi.
Diagnostyka różnicowa polega na badaniu możliwości rozwoju wszystkich tych chorób na podstawie wyników ankiety, badania, badań laboratoryjnych i instrumentalnych.
Najczęściej zespół Tholosa Hunta należy odróżnić od takich patologii:
- zablokowanie zatoki jamistej przez skrzeplinę;
- Zespół Rochona-Duvignota;
- zespół przestrzeni retrospenoidalnej (zespół Jacota);
- zespół paratrigeminal Raedera;
- polineuropatia czaszkowa.
Z kim się skontaktować?
Leczenie zespół Tholosa-Hunta
Zespół Tholosa Hunta dobrze reaguje na leczenie immunosupresyjnym przebiegiem kortykosteroidów hormonalnych. Takie leki są w stanie stłumić agresywną odpowiedź układu odpornościowego i jej niszczący wpływ na tkanki organizmu.
Częściej niż inne leki przepisywane są prednizolon, metyloprednizolon, kortyzon lub alternatywne leki, które wykazały pozytywny wpływ w leczeniu znanych patologii autoimmunologicznych. Wydaje się, że korzyści ze stosowania steroidów są związane z mechanizmem antyoksydacyjnym i / lub zdolnością tak dużych dawek do zmniejszenia obrzęku i późniejszego niedokrwienia w dotkniętym obszarze. [6]
Oprócz kortykosteroidów właściwe jest stosowanie leków przeciwbólowych, przeciwdrgawkowych. Wymagane są kompleksowe preparaty multiwitaminowe.
Jeśli ściśle przestrzegasz wszystkich zaleceń i zaleceń lekarza prowadzącego, bolesne objawy zespołu Tolosa Hunta są szybko zatrzymywane: pacjenci zauważają wyraźną poprawę samopoczucia około drugiego lub trzeciego dnia. W przeważającej większości przypadków zdolność do pracy pozostaje. [7]
Optymalne dawkowanie i częstotliwość przyjmowania leków hormonalnych ustalane są indywidualnie. Nie ma ogólnie przyjętego schematu leczenia, ponieważ bardzo trudno jest zorganizować badania kontrolowane placebo, co wiąże się z niską częstością występowania zespołu. Najczęściej zalecane są wysokie dawki kortykosteroidów, chociaż zdarzały się przypadki skuteczności i raczej małych dawek leków (np. Stosowanie prednizolonu w ilości poniżej 0,5 mg / kg dziennie). Do tej pory średnia ilość prednizolonu stosowanego w zespole Tholosa Hunta wynosi 1-2 mg / kg dziennie.
Przybliżony schemat leczenia:
- Metyloprednizolon (Solu-Medron 1000 jako dożylny wlew kroplowy z 250 ml izotonicznego roztworu chlorku sodu i Panangin (10,0) codziennie przez pięć dni;
- Mildronate do normalizacji metabolizmu komórkowego, dożylne wstrzyknięcie dawki 500 mg dziennie przez 10 dni;
- Neuromidyna poprawiająca przekazywanie impulsów wzdłuż włókien nerwowo-mięśniowych, 20 mg doustnie trzy razy dziennie;
- Clonazepam w celu wzmocnienia działania hamującego na przekazywanie impulsów nerwowych i stymulację receptorów benzodiazepinowych 2 mg doustnie i / lub Trileptal 150 mg doustnie przed snem.
Być może powołanie przedłużonego przebiegu terapii glikokortykosteroidami przy użyciu dużych dawek prednizolonu. [8]
Zapobieganie
Nie można z góry zapobiec pojawieniu się zespołu Tholosa Hunta. Wynika to przynajmniej z faktu, że przyczyny naruszenia nie zostały jeszcze jasno określone. W przypadku stwierdzenia jakichkolwiek bolesnych objawów - w szczególności częstych bólów w okolicy czołowej i oczodołów, podwójnego widzenia i osłabienia mięśni oka, należy jak najszybciej skontaktować się z odpowiednim specjalistą i przeprowadzić pełną diagnostykę.
Profilaktyka wtórna ma na celu zapobieganie nawrotom u pacjentów z rozpoznanym już zespołem Tholosa Hunta. Ważnymi punktami działań zapobiegawczych są:
- regularne konsultacje lekarskie, zabiegi diagnostyczne, nadzór ambulatoryjny specjalistów;
- okresowe kursy terapii kortykosteroidami;
- wzmocnienie i utrzymanie odpowiedniego stanu układu odpornościowego.
Wszyscy pacjenci muszą starać się unikać stresujących sytuacji, aby na czas leczyć wszelkie procesy zapalne w organizmie.
Prognoza
Rokowanie w zespole Tholosa Hunta uważa się za korzystne. Występuje dobra odpowiedź na terapię kortykosteroidami, a samoistna remisja jest powszechna, chociaż niektórzy pacjenci mają skutki resztkowe w postaci upośledzenia funkcji uszkodzonych mięśni oka. W przypadku braku dalszego leczenia choroba nawraca. U leczonych pacjentów nawroty występują w około 35% przypadków. [9]
Po zakończeniu kursu terapeutycznego zwykle przywraca się zdolność do pracy. Jednak odnosi się to do prawidłowo zdiagnozowanej choroby, a nie do innych patologii, które rozwijają się pod „maską” zespołu. [10]
Niepełnosprawność występuje tylko w rzadkich przypadkach. Tylko w przypadku udokumentowanych częstych zaostrzeń można przypisać trzecią grupę niepełnosprawności. W trudnych przypadkach pacjent jest przenoszony na lekki poród, któremu nie towarzyszy stres wizualny. Jeśli zespół Tholosa Hunta jest uporczywy i nawracający, to osobie nie zaleca się prowadzenia pojazdów, co jest spowodowane upośledzoną funkcją motoryczną gałek ocznych i podwójnym widzeniem.