Ekspert medyczny artykułu

Nowe publikacje
Dziedziczne zapalenie nerek (zespół Alporta) u dzieci
Ostatnia recenzja: 05.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
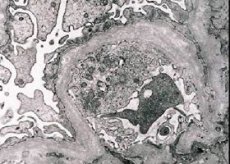
Dziedziczne zapalenie nerek (zespół Alporta) to uwarunkowana genetycznie dziedziczna, nieimmunologiczna kłębuszkowata choroba nerek, objawiająca się krwiomoczem (czasami z białkomoczem), postępującym pogorszeniem czynności nerek z rozwojem przewlekłej niewydolności nerek, często w połączeniu z głuchotą odbiorczą i upośledzeniem wzroku.
Choroba została opisana po raz pierwszy w 1902 roku przez LG Guthrie, który obserwował rodzinę, w której krwiomocz obserwowano w kilku pokoleniach. W 1915 roku AF Hurst opisał rozwój mocznicy u członków tej samej rodziny. W 1927 roku A. Alport po raz pierwszy zidentyfikował utratę słuchu u kilku krewnych z krwiomoczem. W latach 50. XX wieku opisano zmiany oczne w podobnej chorobie. W 1972 roku u pacjentów z dziedziczną krwiomoczem, podczas badania morfologicznego tkanki nerkowej, Hinglais i in. ujawnili nierównomierną ekspansję i stratyfikację błon podstawnych kłębuszków nerkowych. W 1985 roku zidentyfikowano podłoże genetyczne dziedzicznego zapalenia nerek - mutację w genie kolagenu typu IV (Fiengold i in., 1985).
Badanie natury genetycznej choroby pozwoliło nam stwierdzić, że różnice w objawach fenotypowych dziedzicznego zapalenia nerek (z utratą słuchu lub bez) wynikają ze stopnia ekspresji zmutowanego genu. Tak więc obecnie wszystkie warianty kliniczne są uważane za objawy jednej choroby, a termin „dziedziczne zapalenie nerek” jest synonimem terminu „zespół Alporta”.
Badania epidemiologiczne wskazują, że dziedziczne zapalenie nerek występuje z częstością 17 na 100 000 dzieci.
 [
[ Przyczyny zespołu Alporta
Podłoże genetyczne choroby stanowi mutacja w genie łańcucha a-5 kolagenu typu IV. Ten typ jest uniwersalny dla błon podstawnych nerki, aparatu ślimakowego, torebki soczewki, siatkówki i rogówki oka, co zostało udowodnione w badaniach z zastosowaniem przeciwciał monoklonalnych przeciwko tej frakcji kolagenu. Ostatnio wskazano na możliwość wykorzystania sond DNA do diagnostyki prenatalnej dziedzicznego zapalenia nerek.
Podkreśla się znaczenie testowania wszystkich członków rodziny sondami DNA w celu identyfikacji nosicieli zmutowanego genu, co ma ogromne znaczenie w prowadzeniu poradnictwa medycznego i genetycznego rodzin z tą chorobą. Jednak do 20% rodzin nie ma krewnych cierpiących na chorobę nerek, co sugeruje wysoką częstość spontanicznych mutacji nieprawidłowego genu. Większość pacjentów z dziedzicznym zapaleniem nerek ma w swoich rodzinach osoby z chorobą nerek, utratą słuchu i patologią wzroku; małżeństwa spokrewnione między osobami mającymi jednego lub więcej przodków są ważne, ponieważ w małżeństwie osób spokrewnionych prawdopodobieństwo otrzymania tych samych genów od obojga rodziców wzrasta. Ustalono drogi transmisji autosomalne dominujące, autosomalne recesywne i dominujące, sprzężone z chromosomem X.
U dzieci najczęściej rozróżnia się trzy typy dziedzicznego zapalenia nerek: zespół Alporta, dziedziczne zapalenie nerek bez utraty słuchu oraz rodzinną łagodną hematurię.
Zespół Alporta to dziedziczne zapalenie nerek z upośledzeniem słuchu. Opiera się na łącznym defekcie w budowie kolagenu błony podstawnej kłębuszków nerkowych, struktur ucha i oka. Gen klasycznego zespołu Alporta znajduje się w locus 21-22 q długiego ramienia chromosomu X. W większości przypadków dziedziczy się w sposób dominujący, sprzężony z chromosomem X. Pod tym względem zespół Alporta jest cięższy u mężczyzn, ponieważ u kobiet funkcja zmutowanego genu jest kompensowana przez zdrowy allel drugiego, nieuszkodzonego chromosomu.
Podstawą genetyczną rozwoju dziedzicznego zapalenia nerek są mutacje w genach łańcuchów alfa kolagenu typu IV. Znanych jest sześć łańcuchów alfa kolagenu typu IV G: geny łańcuchów a5 i a6 (Col4A5 i Col4A5) znajdują się na długim ramieniu chromosomu X w strefie 21-22q; geny łańcuchów a3 i a4 (Col4A3 i Col4A4) znajdują się na 2. chromosomie; geny łańcuchów a1 i a2 (Col4A1 i Col4A2) znajdują się na 13. chromosomie.
W większości przypadków (80-85%) wykrywa się sprzężony z chromosomem X wzór dziedziczenia choroby, związany z uszkodzeniem genu Col4A5 w wyniku delecji, mutacji punktowych lub zaburzeń splicingu. Obecnie odkryto ponad 200 mutacji genu Col4A5, odpowiedzialnych za zaburzenie syntezy łańcuchów a5 kolagenu typu IV. Przy tym typie dziedziczenia choroba objawia się u dzieci obojga płci, ale u chłopców jest cięższa.
Mutacje w loci genów Col4A3 i Col4A4 odpowiedzialnych za syntezę łańcuchów a3 i a4 kolagenu typu IV są dziedziczone autosomalnie. Według badań, typ dziedziczenia autosomalnego dominującego obserwuje się w 16% przypadków dziedzicznego zapalenia nerek, a typ autosomalny recesywny obserwuje się u 6% pacjentów. Znanych jest około 10 wariantów mutacji genów Col4A3 i Col4A4.
Skutkiem mutacji jest naruszenie procesów montażu kolagenu typu IV, co prowadzi do naruszenia jego struktury. Kolagen typu IV jest jednym z głównych składników błony podstawnej kłębuszków nerkowych, aparatu ślimakowego i soczewki oka, którego patologię można wykryć w klinice dziedzicznego zapalenia nerek.
Kolagen typu IV, który jest częścią błony podstawnej kłębuszków nerkowych, składa się głównie z dwóch łańcuchów a1 (IV) i jednego łańcucha a2 (IV), a także zawiera łańcuchy a3, a4, a5. Najczęściej w dziedziczeniu sprzężonym z chromosomem X mutacja genu Col4A5 jest powiązana z brakiem łańcuchów a3, a4, a5 i a6 w strukturze kolagenu typu IV, a liczba łańcuchów o1 i a2 w błonie podstawnej kłębuszków nerkowych wzrasta. Mechanizm tego zjawiska jest niejasny, przyjmuje się, że przyczyną są zmiany potranskrypcyjne w mRNA.
Brak łańcuchów a3, a4 i a5 w strukturze kolagenu typu IV błon podstawnych kłębuszków nerkowych prowadzi do ich ścieńczenia i kruchości we wczesnych stadiach zespołu Alporta, co klinicznie objawia się częściej krwiomoczem (rzadziej krwiomoczem z białkomoczem lub tylko białkomoczem), utratą słuchu i stożkiem soczewkowym. Dalszy postęp choroby prowadzi do pogrubienia i upośledzenia przepuszczalności błon podstawnych w późnych stadiach choroby, z proliferacją w nich kolagenów typu V i VI, objawiającą się wzrostem białkomoczu i spadkiem czynności nerek.
Charakter mutacji leżącej u podłoża dziedzicznego zapalenia nerek w dużej mierze determinuje jego manifestację fenotypową. W przypadku delecji chromosomu X z jednoczesną mutacją genów Col4A5 i Col4A6 odpowiedzialnych za syntezę łańcuchów a5 i a6 kolagenu typu IV, zespół Alporta łączy się z leiomiomatozą przełyku i narządów płciowych. Według danych badawczych, w przypadku mutacji genu Col4A5 związanej z delecją, obserwuje się większe nasilenie procesu patologicznego, połączenie uszkodzenia nerek z objawami pozanerkowymi i wczesnym rozwojem przewlekłej niewydolności nerek, w porównaniu z mutacją punktową tego genu.
Morfologicznie, mikroskopia elektronowa ujawnia ścieńczenie i rozwarstwienie błon podstawnych kłębuszków nerkowych (zwłaszcza blaszki gęstej) oraz obecność gęstych elektronowo granulek. Zmiany kłębuszkowe mogą być niejednorodne u tego samego pacjenta, od minimalnych ogniskowych zmian mezangialnych do stwardnienia kłębuszków nerkowych. Zapalenie kłębuszków nerkowych w zespole Alporta jest zawsze immunoujemne, co odróżnia je od kłębuszkowego zapalenia nerek. Charakterystyczne cechy obejmują rozwój zaniku kanalikowego, naciek limfohistiocytarny i obecność „komórek piankowatych” z inkluzjami lipidowymi - lipofagów. W miarę postępu choroby ujawnia się pogrubienie i wyraźne zniszczenie błon podstawnych kłębuszków nerkowych.
Ujawniają się pewne zmiany w układzie odpornościowym. U pacjentów z dziedzicznym zapaleniem nerek występuje obniżony poziom IgA i tendencja do wzrostu stężenia IgM we krwi, poziom IgG może być podwyższony we wczesnych stadiach choroby i obniżony w późniejszych stadiach. Być może wzrost stężenia IgM i G jest rodzajem reakcji kompensacyjnej w odpowiedzi na niedobór IgA.
Zmniejsza się aktywność funkcjonalna układu limfocytów T, obserwuje się selektywne zmniejszenie limfocytów B odpowiedzialnych za syntezę IgA, zaburzone zostaje powiązanie fagocytarne odporności, głównie na skutek zaburzenia procesów chemotaksji i trawienia wewnątrzkomórkowego w neutrofilach
Podczas badania biopsji nerki u pacjentów z zespołem Alporta dane z mikroskopii elektronowej ujawniają zmiany ultrastrukturalne w błonie podstawnej kłębuszków nerkowych: ścieńczenie, zaburzenie struktury i rozszczepienie błon podstawnych kłębuszków nerkowych ze zmianą ich grubości i nierównymi konturami. We wczesnych stadiach dziedzicznego zapalenia nerek defekt determinuje ścieńczenie i kruchość błon podstawnych kłębuszków nerkowych.
Ścieńczenie błon kłębuszkowych jest korzystniejszym objawem i występuje częściej u dziewcząt. Bardziej stałym objawem mikroskopowym w dziedzicznym zapaleniu nerek jest rozszczepienie błony podstawnej, a stopień jej zniszczenia koreluje z ciężkością procesu.
Objawy zespołu Alporta u dzieci
Pierwsze objawy zespołu Alporta w postaci izolowanego zespołu moczowego są najczęściej wykrywane u dzieci w pierwszych trzech latach życia. W większości przypadków choroba jest wykrywana przypadkowo. Zespół moczowy jest wykrywany podczas badania profilaktycznego dziecka, przed przyjęciem do placówki opiekuńczej lub w trakcie ARVI. W przypadku patologii w moczu w trakcie ARVI. W dziedzicznym zapaleniu nerek, w przeciwieństwie do nabytego kłębuszkowego zapalenia nerek, nie ma okresu utajonego.
W początkowym stadium choroby zdrowie dziecka cierpi w niewielkim stopniu, charakterystyczną cechą jest uporczywość i oporność zespołu moczowego. Jednym z głównych objawów jest krwiomocz o różnym stopniu nasilenia, obserwowany w 100% przypadków. Wzrost stopnia krwiomoczu obserwuje się w trakcie lub po infekcjach układu oddechowego, aktywności fizycznej lub po szczepieniach profilaktycznych. Białkomocz w większości przypadków nie przekracza 1 g/dobę, na początku choroby może być niestały, w miarę postępu procesu białkomocz wzrasta. Okresowo w osadzie moczu może występować leukocyturia z przewagą limfocytów, co wiąże się z rozwojem zmian śródmiąższowych.
Następnie dochodzi do częściowego upośledzenia czynności nerek, pogarsza się ogólny stan pacjenta: zatrucie, osłabienie mięśni, niedociśnienie tętnicze, często upośledzenie słuchu (szczególnie u chłopców), a czasem pojawiają się zaburzenia widzenia. Zatrucie objawia się bladością, zmęczeniem i bólami głowy. W początkowym stadium choroby utrata słuchu jest w większości przypadków wykrywana tylko za pomocą audiografii. Utrata słuchu w zespole Alporta może wystąpić w różnych okresach dzieciństwa, ale najczęściej utratę słuchu diagnozuje się w wieku 6-10 lat. Utrata słuchu u dzieci zaczyna się od wysokich częstotliwości, osiągając znaczny stopień w przewodnictwie powietrznym i kostnym, przechodząc od ubytku słuchu przewodzącego dźwięk do ubytku słuchu percepcyjnego. Utrata słuchu może być jednym z pierwszych objawów choroby i może poprzedzać zespół moczowy.
U 20% pacjentów z zespołem Alporta występują zmiany w narządach wzroku. Najczęściej wykrywanymi anomaliami są nieprawidłowości soczewki: sferofokia, przedni, tylny lub mieszany stożek soczewki oraz różne zaćmy. W rodzinach z zespołem Alporta występuje znaczna częstość krótkowzroczności. Wielu badaczy stale odnotowuje obustronne zmiany okołoplamkowe u tych rodzin w postaci jasnych białawych lub żółtawych ziarnistości w ciałku żółtym. Uważają ten objaw za stały objaw, który ma wysoką wartość diagnostyczną w zespole Alporta. KS Chugh i in. (1993) w badaniu okulistycznym stwierdzili u pacjentów z zespołem Alporta pogorszenie ostrości wzroku w 66,7% przypadków, przedni stożek soczewki w 37,8%, plamy siatkówkowe w 22,2%, zaćmę w 20% i stożek rogówki w 6,7%.
U niektórych dzieci z dziedzicznym zapaleniem nerek, zwłaszcza gdy rozwija się niewydolność nerek, zauważa się znaczne opóźnienie w rozwoju fizycznym. Wraz z postępem niewydolności nerek rozwija się nadciśnienie tętnicze. U dzieci jest ono częściej wykrywane w okresie dojrzewania i w starszych grupach wiekowych.
U pacjentów z dziedzicznym zapaleniem nerek występuje wiele (ponad 5-7) znamion dysmorfogenezy tkanki łącznej. Wśród znamion tkanki łącznej u pacjentów najczęściej występują hiperteloryzm oczu, wysokie podniebienie, anomalie zgryzu, nieprawidłowy kształt małżowin usznych, krzywizna małego palca u rąk i „przerwa sandałowa” u stóp. Dziedziczne zapalenie nerek charakteryzuje się jednolitością znamion dysmorfogenezy w obrębie rodziny, a także wysoką częstością ich występowania wśród krewnych probantów, w linii których choroba jest przenoszona.
We wczesnych stadiach choroby obserwuje się izolowane zmniejszenie częściowych funkcji nerek: transportu aminokwasów, elektrolitów, funkcji koncentracji, kwasogenezy, późniejsze zmiany dotyczą stanu czynnościowego zarówno części proksymalnej, jak i dystalnej nefronu i charakteryzują się łączonymi zaburzeniami częściowymi. Spadek filtracji kłębuszkowej występuje później, częściej w okresie dojrzewania. W miarę postępu dziedzicznego zapalenia nerek rozwija się niedokrwistość.
W ten sposób dziedziczne zapalenie nerek charakteryzuje się etapowym przebiegiem choroby: najpierw utajone stadium lub ukryte objawy kliniczne, objawiające się minimalnymi zmianami w zespole moczowym, następnie następuje stopniowa dekompensacja procesu ze spadkiem funkcji nerek z wyraźnymi objawami klinicznymi (zatrucie, osłabienie, opóźnienie rozwoju, niedokrwistość). Objawy kliniczne pojawiają się zazwyczaj niezależnie od warstwowości reakcji zapalnej.
Dziedziczne zapalenie nerek może ujawnić się w różnym okresie wieku, co zależy od działania genu, który do pewnego czasu znajduje się w stanie represji.
Klasyfikacja
Istnieją trzy rodzaje dziedzicznego zapalenia nerek
- Wariant I - klinicznie objawia się zapaleniem nerek z krwiomoczem, utratą słuchu i uszkodzeniem oczu. Przebieg zapalenia nerek jest postępujący, z rozwojem przewlekłej niewydolności nerek. Typ dziedziczenia jest dominujący, związany z chromosomem X. Morfologicznie ujawnia się naruszenie struktury błony podstawnej, jej ścieńczenie i rozszczepienie.
- Opcja II - klinicznie objawia się zapaleniem nerek z krwiomoczem bez utraty słuchu. Przebieg zapalenia nerek jest postępujący wraz z rozwojem przewlekłej niewydolności nerek. Typ dziedziczenia jest dominujący, związany z chromosomem X. Morfologicznie stwierdza się ścieńczenie błony podstawnej naczyń włosowatych kłębuszków nerkowych (zwłaszcza laminadensa).
- Opcja III - łagodna rodzinna hematuria. Przebieg jest korzystny, przewlekła niewydolność nerek nie rozwija się. Typ dziedziczenia jest autosomalny dominujący lub autosomalny recesywny. Przy autosomalnym recesywnym typie dziedziczenia u kobiet obserwuje się cięższy przebieg choroby.
Diagnoza zespołu Alporta
Proponowane są następujące kryteria:
- obecność co najmniej dwóch pacjentów z nefropatią w każdej rodzinie;
- krwiomocz jako główny objaw nefropatii u probanta;
- występowanie ubytku słuchu u co najmniej jednego członka rodziny;
- rozwój przewlekłej niewydolności nerek u jednego lub kilku krewnych.
W diagnostyce różnych chorób dziedzicznych i wrodzonych duże miejsce zajmuje kompleksowe podejście do badania i przede wszystkim zwrócenie uwagi na dane uzyskane przy sporządzaniu rodowodu dziecka. Rozpoznanie zespołu Alporta uważa się za zasadne w przypadkach, gdy u pacjenta stwierdza się 3 z 4 typowych objawów: obecność krwiomoczu i przewlekłej niewydolności nerek w rodzinie, obecność niedosłuchu neurosensorycznego, patologii widzenia u pacjenta, wykrycie objawów rozszczepienia błony podstawnej kłębuszków nerkowych ze zmianą jej grubości i nierównymi konturami podczas charakterystyki mikroskopowej biopsji elektronowej.
Badanie pacjenta powinno obejmować kliniczne i genetyczne metody badawcze; ukierunkowane badanie historii choroby; ogólne badanie pacjenta z uwzględnieniem diagnostycznie istotnych kryteriów. W fazie kompensacyjnej patologię można wykryć tylko poprzez skupienie się na takich zespołach, jak obecność obciążenia dziedzicznego, niedociśnienie, liczne znamiona dysembriogenezy, zmiany w zespole moczowym. W fazie dekompensacji mogą pojawić się objawy pozanerkowe, takie jak ciężkie zatrucie, astenia, opóźniony rozwój fizyczny, niedokrwistość, objawiające się i nasilające wraz ze stopniowym spadkiem czynności nerek. U większości pacjentów wraz ze spadkiem czynności nerek obserwuje się: zmniejszenie kwaso- i aminogenezy; u 50% pacjentów obserwuje się znaczne zmniejszenie czynności wydzielniczej nerek; ograniczony zakres wahań gęstości optycznej moczu; zaburzenie rytmu filtracji, a następnie zmniejszenie filtracji kłębuszkowej. Stopień przewlekłej niewydolności nerek rozpoznaje się, gdy u pacjenta utrzymuje się podwyższone stężenie mocznika w surowicy krwi (powyżej 0,35 g/l) przez okres 3-6 miesięcy lub dłużej i następuje spadek filtracji kłębuszkowej do 25% normy.
Diagnostykę różnicową dziedzicznego zapalenia nerek należy przeprowadzić przede wszystkim w przypadku postaci krwiomoczowej nabytego kłębuszkowego zapalenia nerek. Nabyte kłębuszkowe zapalenie nerek ma najczęściej ostry początek, okres 2-3 tygodni po zakażeniu, objawy pozanerkowe, w tym nadciśnienie od pierwszych dni (w dziedzicznym zapaleniu nerek przeciwnie, niedociśnienie), obniżoną filtrację kłębuszkową na początku choroby, brak upośledzenia częściowych funkcji kanalikowych, podczas gdy w dziedzicznym są one obecne. Nabyte kłębuszkowe zapalenie nerek przebiega z wyraźniejszą krwiomoczem i białkomoczem, ze zwiększonym OB. Typowe zmiany w błonie podstawnej kłębuszków, charakterystyczne dla dziedzicznego zapalenia nerek, mają wartość diagnostyczną.
Diagnostyka różnicowa z nefropatią dysmetaboliczną przeprowadzana jest przy przewlekłej niewydolności nerek, w rodzinie klinicznie ujawnionych heterogenicznych chorobach nerek, a może występować spektrum nefropatii od odmiedniczkowego zapalenia nerek do kamicy moczowej. Dzieci często skarżą się na bóle w podbrzuszu i okresowo podczas oddawania moczu, w osadzie moczu - szczawiany.
W przypadku podejrzenia dziedzicznego zapalenia nerek należy skierować pacjenta do specjalistycznego oddziału nefrologicznego w celu wyjaśnienia rozpoznania.
Co trzeba zbadać?
Jakie testy są potrzebne?
Z kim się skontaktować?
Leczenie zespołu Alporta
Schemat obejmuje ograniczenia dużego wysiłku fizycznego i przebywania na świeżym powietrzu. Dieta jest kompletna, z odpowiednim poziomem pełnowartościowych białek, tłuszczów i węglowodanów, uwzględniająca funkcję nerek. Duże znaczenie ma wykrywanie i leczenie przewlekłych ognisk infekcji. Stosowane są następujące leki: ATP, kokarboksylaza, pirydoksyna (do 50 mg/dobę), chlorek karnityny. Kursy są podawane 2-3 razy w roku. Na krwiomocz przepisuje się ziołolecznictwo - pokrzywę, sok z aronii, krwawnik.
Istnieją doniesienia w literaturze zagranicznej i krajowej o leczeniu prednizolonem i stosowaniu cytostatyków. Trudno jednak ocenić efekt.
W przewlekłej niewydolności nerek stosuje się hemodializę i przeszczep nerki.
Nie ma metod specyficznej (skutecznej patogenetycznej) terapii dziedzicznego zapalenia nerek. Wszystkie środki lecznicze mają na celu zapobieganie i spowolnienie spadku funkcji nerek.
Dieta powinna być zrównoważona i wysokokaloryczna, uwzględniając stan czynnościowy nerek. W przypadku braku zaburzeń czynnościowych dieta dziecka powinna zawierać wystarczającą ilość białek, tłuszczów i węglowodanów. W przypadku występowania objawów dysfunkcji nerek należy ograniczyć ilość białka, węglowodanów, wapnia i fosforu, co opóźnia rozwój przewlekłej niewydolności nerek.
Należy ograniczyć aktywność fizyczną; dzieciom zaleca się unikanie uprawiania sportu.
Należy unikać kontaktu z pacjentami zakaźnymi, należy ograniczyć ryzyko wystąpienia ostrych chorób układu oddechowego. Konieczna jest dezynfekcja ognisk przewlekłego zakażenia. U dzieci z dziedzicznym zapaleniem nerek nie wykonuje się szczepień profilaktycznych, szczepienie jest możliwe jedynie ze wskazań epidemiologicznych.
Terapia hormonalna i immunosupresyjna w dziedzicznym zapaleniu nerek jest nieskuteczna. Istnieją przesłanki wskazujące na pewien pozytywny efekt (zmniejszenie białkomoczu i spowolnienie postępu choroby) przy długotrwałym, wieloletnim stosowaniu cyklosporyny A i inhibitorów ACE.
W leczeniu pacjentów stosuje się leki poprawiające metabolizm:
- pirydoksyna - 2-3 mg/kg/dobę w 3 dawkach przez 4 tygodnie;
- kokarboksylaza - 50 mg domięśniowo co drugi dzień, łącznie 10-15 zastrzyków;
- ATP - 1 ml domięśniowo co drugi dzień, 10-15 zastrzyków;
- witamina A - 1000 IU/rok/dzień w 1 dawce przez 2 tygodnie;
- Witamina E - 1 mg/kg/dzień w 1 dawce przez 2 tygodnie.
Ten rodzaj terapii pomaga poprawić ogólny stan chorych, zmniejszyć dysfunkcje kanalikowe i przeprowadza się ją w seriach 3 razy w roku.
Lewamizol można stosować jako immunomodulator - 2 mg/kg/dobę 2-3 razy w tygodniu z przerwami między dawkami wynoszącymi 3-4 dni.
Dane naukowe dowodzą, że tlenoterapia hiperbaryczna ma pozytywny wpływ na nasilenie krwiomoczu i dysfunkcji nerek.
Najskuteczniejszą metodą leczenia dziedzicznego zapalenia nerek jest terminowy przeszczep nerki. W tym przypadku nie dochodzi do nawrotu choroby w przeszczepie; w niewielkim procencie przypadków (około 5%) może rozwinąć się zapalenie nerek w przeszczepionej nerce związane z antygenami błony podstawnej kłębuszków nerkowych.
Obiecującym kierunkiem jest diagnostyka prenatalna i terapia inżynierii genetycznej. Eksperymenty na zwierzętach wykazują wysoką skuteczność transferu normalnych genów odpowiedzialnych za syntezę łańcuchów alfa kolagenu typu IV do tkanki nerkowej, po czym obserwuje się syntezę normalnych struktur kolagenowych.
Prognoza
Rokowanie w przypadku dziedzicznego zapalenia nerek jest zawsze poważne.
Kryteriami prognostycznie niekorzystnymi dla przebiegu dziedzicznego zapalenia nerek są:
- płeć męska;
- wczesny rozwój przewlekłej niewydolności nerek u członków rodziny;
- białkomocz (ponad 1 g/dobę);
- pogrubienie błon podstawnych kłębuszków nerkowych widoczne w badaniu mikroskopowym;
- zapalenie nerwu słuchowego;
- usunięcie w genie Col4A5.
Rokowanie w przypadku łagodnej rodzinnej hematurii jest korzystniejsze.
Использованная литература