Ekspert medyczny artykułu

Nowe publikacje
Rola zmian w kości podchrzęstnej w patogenezie choroby zwyrodnieniowej stawów
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
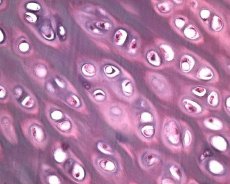
Wraz z degeneracją chrząstki stawowej, w proces patologiczny w osteoartrozie zaangażowana jest również tkanka kostna. Zakłada się, że pogrubienie płytki podchrzęstnej przyczynia się do postępu osteoartrozy. W miarę postępu osteoartrozy chrząstka stawowa, która jest poddawana obciążeniom mechanicznym i chemicznym, powoli ulega erozji z powodu braku równowagi w procesach katabolizmu i naprawy chrząstki. W szczególności obciążenie mechaniczne w stosunku do stawów „nośnych” ciężaru ciała przyczynia się do powstawania dużej liczby mikropęknięć w płytce podchrzęstnej i chrząstce. W miarę erozji chrząstki stawowej postępuje stwardnienie kości podchrzęstnej, wzrasta sztywność tkanki kostnej, co z kolei przyczynia się do dalszego zaburzenia struktury chrząstki stawowej. Jednak kwestia pierwotnej lub wtórnej natury zmian kości podchrzęstnej w osteoartrozie pozostaje nierozwiązana.
Do niedawna zmiany w gąbczastej substancji kości podchrzęstnej, takie jak stwardnienie lub tworzenie torbieli, wykrywalne radiologicznie, były uważane za wtórne u pacjentów z chorobą zwyrodnieniową stawów. Jednak wyniki badań klinicznych i eksperymentalnych wskazują na możliwą inicjującą rolę kości podchrzęstnej w patogenezie choroby zwyrodnieniowej stawów. Jednym z możliwych mechanizmów jest gwałtowny wzrost gradientu sztywności kości podchrzęstnej z powodu faktu, że integralność leżącej pod nią tkanki chrzęstnej zależy od właściwości mechanicznych jej „łożyska” kostnego. Badania na naczelnych wykazały, że zmiany w kości podchrzęstnej mogą poprzedzać zmiany w chrząstce stawowej. Dowody za i przeciw tej hipotezie, które pojawiły się w wyniku badań na zwierzęcych modelach choroby zwyrodnieniowej stawów i badań klinicznych, tylko zaostrzyły debatę. Pogrubienie beleczek w kości podchrzęstnej nie zawsze towarzyszy wzrostowi mineralizacji kości, a raczej wzrostowi objętości osteoidu. Ten objaw nieprawidłowej mineralizacji wskazuje, że zaburzenie regulacji przebudowy kości jest integralną częścią choroby zwyrodnieniowej stawów i również wspiera koncepcję defektu komórek kostnych w chorobie zwyrodnieniowej stawów. Grupa J. Dequekera (1989) uważa tę ostatnią za „uogólnioną metaboliczną chorobę kości”.
Tkanka kostna jest stale odnawiana. Ten dynamiczny proces, zwany przebudową kości, jest złożoną sekwencją resorpcji i mineralizacji. Osteoklasty resorbują tkankę kostną, a osteoblasty wydzielają białka, które stanowią główny składnik organiczny do mineralizacji. Tworzenie kości i resorpcja nie zachodzą losowo w całym szkielecie; jest to zaprogramowany proces, który zachodzi w różnych obszarach szkieletu, zwanych jednostkami przebudowy kości. Na początku cyklu osteoklasty pojawiają się na nieaktywnej powierzchni; w ciągu 2 tygodni tworzą tunel w kości korowej lub lukę na powierzchni kości beleczkowej. Częstotliwość aktywacji nowych jednostek przebudowy kości determinuje stopień odnowy kości. U zdrowej młodej osoby procesy tworzenia i resorpcji kości są zrównoważone, a normalna masa kostna jest utrzymywana. W hormonalnej regulacji resorpcji tkanki kostnej, przynajmniej PTH i PGE2 , uczestniczą nie tylko osteoklasty, ale także osteoblasty, ponieważ pod wpływem tych hormonów uwalniane są czynniki stymulujące resorpcję kości przez osteoklasty. Obecnie znanych jest ponad 12 lokalnych i systemowych regulatorów wzrostu tkanki kostnej, które wpływają na jej przebudowę. W szczególności należą do nich: PTH, 1,25(OH) 2D3 ,kalcytonina, hormon wzrostu, glikokortykoidy, hormony tarczycy, insulina, IGF (1 i 2), estrogeny, PGE2 , androgeny.
Komórki kostne uwalniają szereg białek i cytokin, które regulują gospodarkę hormonalną i przekazują sygnały. Białka wytwarzane przez osteoblasty obejmują białka macierzy kostnej, takie jak kolagen, osteopontyna, osteokalcyna, sialoproteiny kostne. Ponadto komórki te uwalniają proteazy w formach aktywnych i utajonych, które uczestniczą w procesie przebudowy tkanki kostnej - MMP, składniki układu aktywatora plazminogenu (PA)/plazminy. Cytokiny uwalniane przez osteoblasty mogą działać zarówno poprzez mechanizmy autokrynne, jak i parakrynne na komórki lokalne (inne osteoblasty, osteoklasty).
Nie wiadomo jeszcze, czy sygnały te są regulowane przez stres mechaniczny lub inne sygnały chemiczne indukowane przez stres mechaniczny. Wiadomo jednak, że powtarzający się stres mechaniczny powoduje lokalną proliferację komórek kostnych i/lub białek. In vivo obciążenie mechaniczne może aktywować osteoblasty, zwiększać poziom cyklicznych nukleotydów, produkcję prostaglandyn i powodować zmiany morfologiczne związane z przebudową kości. In vitro stres mechaniczny powoduje proliferację kultur osteoblastów, ekspresję mRNA białek kostnych zaangażowanych w formowanie i mineralizację osteoidów, uwalnianie lokalnych czynników wzrostu, takich jak IGF-1 i IGF-2, oraz cząsteczek adhezyjnych. Transmisja sygnału stresu mechanicznego może odbywać się za pośrednictwem kanałów jonowych wrażliwych na działanie mechano.
Istnieją pośrednie dowody na dysfunkcję osteoblastów w osteoartrozie. G. Gevers i J. Dequeker (1987) wykazali wzrost poziomu osteokalcyny w surowicy u kobiet z osteoartrozą dłoni, jak również w przypadku wycinków kości korowej, co wskazuje, że patologia kości może być częścią osteoartrozy. Autopsja wykazała nie tylko pogrubienie kości podchrzęstnej, ale także nieprawidłowo niską mineralizację głowy kości udowej. U świnek morskich z osteoartrozą wywołaną chirurgicznie, tomografia komputerowa wykazała znaczne pogrubienie frakcji kostnej w strefie podchrzęstnej. Nierównowaga między białkami kolagenowymi i niekolagenowymi (osteokalcyna itp.) może prowadzić do zwiększenia objętości kości, ale nie wpływa na jej gęstość mineralną. Według M. Shimizu i in. (1993) postęp zmian zwyrodnieniowych w chrząstce stawowej wiąże się z intensywniejszą przebudową kości podchrzęstnej i wzrostem jej sztywności, co również wskazuje na defekt komórek tkanki kostnej w osteoartrozie. Zgodnie z hipotezą zaproponowaną przez B. Lee i M. Aspdena (1997) proliferacja wadliwych komórek kostnych może prowadzić do zwiększenia sztywności tkanki kostnej, ale nie powoduje zwiększenia jej gęstości mineralnej.
CI Westacott i in. (1997) wysunęli hipotezę, że nieprawidłowe osteoblasty bezpośrednio wpływają na metabolizm chrząstki. Hodując osteoblasty od pacjentów z chorobą zwyrodnieniową stawów z chondrocytami od osób, które nie miały chorób stawów, autorzy zaobserwowali znaczącą zmianę w uwalnianiu glikozaminoglikanów przez normalną tkankę chrzęstną in vitro, ale poziom uwalniania cytokin pozostał niezmieniony. G. Hilal i in. (1998) wykazali, że hodowla osteoblastów z kości podchrzęstnej pacjentów z chorobą zwyrodnieniową stawów in vitro ma zmieniony metabolizm - aktywność układu AP/plazmina i poziom IGF-1 w tych komórkach są zwiększone. Obserwację CI Westacott i in. (1997) można wyjaśnić wzrostem aktywności proteaz w komórkach kości podchrzęstnej.
Nie wiadomo, czy zmiany w kości podchrzęstnej inicjują chorobę zwyrodnieniową stawów, czy przyczyniają się do jej postępu. DK Dedrick i in. (1993) wykazali, że u psów z wywołaną chirurgicznie chorobą zwyrodnieniową stawów pogrubienie kości podchrzęstnej nie jest warunkiem koniecznym do rozwoju zmian podobnych do choroby zwyrodnieniowej stawów w chrząstce stawowej, ale przyczynia się do postępu procesów zwyrodnieniowych w chrząstce. Wyniki badania przeprowadzonego przez A. Sa'ied i in. (1997) przeczą danym z poprzedniego badania. Wykorzystując echografię 50 MHz do oceny początkowych zmian morfologicznych i ich postępu w chrząstce stawowej i kości w eksperymentalnej chorobie zwyrodnieniowej stawów wywołanej wstrzyknięciami kwasu monojodooctowego do stawu kolanowego szczurów, autorzy wykazali jednoczesny proces zmian w kości i chrząstce w ciągu pierwszych trzech dni po wstrzyknięciu.
Osteoblasty wydzielają czynniki wzrostu i cytokiny biorące udział w miejscowej przebudowie kości, które mogą promować przebudowę właściwej chrząstki w stawach „przenoszących ciężar” po ich przeniknięciu przez mikropęknięcia w zwapniałej warstwie chrząstki stawowej. Ponadto produkty wydzielnicze komórek kostnych znajdują się w płynie stawowym. Najbardziej prawdopodobnymi produktami wydzielanymi przez nieprawidłowe osteoblasty, które mogą inicjować proces miejscowej przebudowy chrząstki, są TGF-b i białka morfometrii kości (BMP). Obaj członkowie rodziny TGF są wydzielani zarówno przez chondrocyty, jak i osteoblasty, a oba są zdolne do modyfikowania przebudowy zarówno kości, jak i chrząstki. J. Martel Pelletier i in. (1997) zaobserwowali wzrost poziomu TGF-β w podchrzęstnych eksplantatach kości u pacjentów z chorobą zwyrodnieniową stawów w porównaniu do osób zdrowych, co wskazuje na prawdopodobną rolę tego czynnika wzrostu w patogenezie choroby zwyrodnieniowej stawów. IGF są również produkowane przez osteoblasty. W hodowli komórek osteoblastycznych pobranych od pacjentów cierpiących na chorobę zwyrodnieniową stawów stwierdzono wzrost poziomu IGF-ów, które zmieniają metabolizm chrząstki.
TGF-b, IGF, BMP i cytokiny wytwarzane przez osteoblasty w kości podchrzęstnej mogą wpływać na produkcję kolagenazy i innych enzymów proteolitycznych w chrząstce, co z kolei może promować przebudowę/degradację macierzy chrząstki. Nadal nie jest jasne, czy osteoblasty w OA produkują mniej czynnika stymulującego kolonie makrofagów (M-CSF - stymulatora resorpcji kości) niż normalne komórki. Wyniki badań AG Uitterlinden i in. (1997) wykazały, że receptory witaminy D, które są wyrażane przez osteoblasty i regulują ekspresję szeregu czynników syntetyzowanych przez te komórki, mogą odgrywać pewną rolę w tworzeniu osteofitów, co częściowo wyjaśnia rolę osteoblastów w patogenezie tej choroby.
Biorąc pod uwagę wyniki powyższych badań, G. Hilal i in. (1998), J. Martel-Pelletier i in. (1997) zaproponowali następującą hipotezę roboczą dotyczącą związku między przebudową kości podchrzęstnej a prawidłową chrząstką stawową w chorobie zwyrodnieniowej stawów. Na wczesnym lub zaawansowanym etapie patogenezy OA proces przebudowy tkanki kostnej w kości podchrzęstnej nasila się. Jednocześnie powtarzające się obciążenia prowadzą do lokalnych mikropęknięć i/lub pojawienia się nierównowagi w układzie IGF/białko wiążące IGF (IGFBP) z powodu nieprawidłowej odpowiedzi osteoblastów kości podchrzęstnej, co przyczynia się do jej stwardnienia. To ostatnie z kolei może przyczyniać się do pojawienia się mikropęknięć prawidłowej chrząstki i uszkodzenia jej macierzy.
W normalnych warunkach uszkodzenie to jest naprawiane przez lokalną syntezę i uwalnianie IGF-1 oraz białka wiążącego IGF, które stymulują tworzenie ECM chrząstki stawowej. Jednocześnie układ GF promuje wzrost komórek kości podchrzęstnej i tworzenie macierzy kostnej. Aktywność anaboliczna układu IGF jest zwiększona w kości podchrzęstnej pacjentów z chorobą zwyrodnieniową stawów, podczas gdy lokalna aktywacja układu AP/plazminy (lokalnego regulatora układu IGF) w chrząstce stawowej powoduje jej lokalne zmiany. W osteoblastach w chorobie zwyrodnieniowej stawów IGF-1 zakłóca regulację AP przez plazminę za pomocą dodatniego sprzężenia zwrotnego, dlatego może powstrzymać przebudowę tkanki kostnej, co ostatecznie prowadzi do stwardnienia podchrzęstnego. Tak więc w tkance kostnej i chrzęstnej lokalna indukcja IGF-1 i proteaz prowadzi z jednej strony do uszkodzenia chrząstki, z drugiej strony do pogrubienia kości podchrzęstnej, co z kolei przyczynia się do dalszego uszkodzenia chrząstki. Nierównowaga między uszkodzeniem chrząstki związanym ze stwardnieniem podchrzęstnym a jej zdolnościami naprawczymi prowadzi do postępujących zmian w ECM chrząstki i do rozwoju osteoartrozy. Według autorów hipoteza ta wyjaśnia również powolny postęp choroby.