Ekspert medyczny artykułu

Nowe publikacje
Zespół Apera
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.

 [
[ Przyczyny Zespół Apera
Rozwój zespołu jest wywoływany przez zaburzenie w budowie genu zlokalizowanego na chromosomie 10. Gen ten odpowiada za proces rozłączania palców, a ponadto za terminowe zamykanie szwów czaszkowych.
Ponadto za przyczynę patologii uważa się choroby zakaźne przebyte w czasie ciąży (takie jak różyczka lub gruźlica, kiła lub grypa, a także zapalenie opon mózgowych) oraz narażenie matki na promieniowanie rentgenowskie. Najczęściej taki zespół wykrywa się u dzieci urodzonych przez rodziców w podeszłym wieku.
Patogeneza
Zespół Aperta jest dziedziczny. Jego typ jest autosomalny dominujący (czyli w rodzinie, w której jedno z przyszłych rodziców jest chore, prawdopodobieństwo urodzenia dziecka z tą chorobą wynosi 50-100%).
Unikalna mutacja w receptorze czynnika wzrostu fibroblastów 2 (FGFR2) powoduje zwiększoną liczbę komórek progenitorowych, które rozwijają się wzdłuż ścieżki osteogenicznej. Ostatecznie prowadzi to do zwiększonego tworzenia podokostnowej macierzy kostnej i przedwczesnego kostnienia szwów czaszkowych w trakcie rozwoju płodowego. Kolejność i szybkość topnienia szwów determinują stopień deformacji i niepełnosprawności. Po wygojeniu się materiału szwu wzrost innych tkanek prostopadle do tego szwu jest ograniczony, a zrośnięte kości działają jak pojedyncze rusztowanie kostne.
Pierwszym dowodem genetycznym na to, że syndaktylia w zespole Aperta jest spowodowana defektem receptora czynnika wzrostu keratynocytów (KGFR), była obserwacja korelacji między ekspresją KGFR w fibroblastach a ciężkością syndaktylii.
Niedowidzenie i zez są częstsze u pacjentów z mutacją FGFR2 Ser252Trp, a zanik tarczy nerwu wzrokowego jest częstszy u pacjentów z mutacją FGFR2 Pro253Arg. U pacjentów z mutacjami FGR2 Ser252Trp częstość występowania upośledzenia wzroku jest znacznie wyższa w porównaniu do pacjentów z mutacją FGFR2 Pro253Arg.
Objawy Zespół Apera
Niektóre objawy choroby są wyraźnie widoczne już przy narodzinach dziecka, ponieważ rozwijają się w łonie matki. Do głównych objawów zespołu należą:
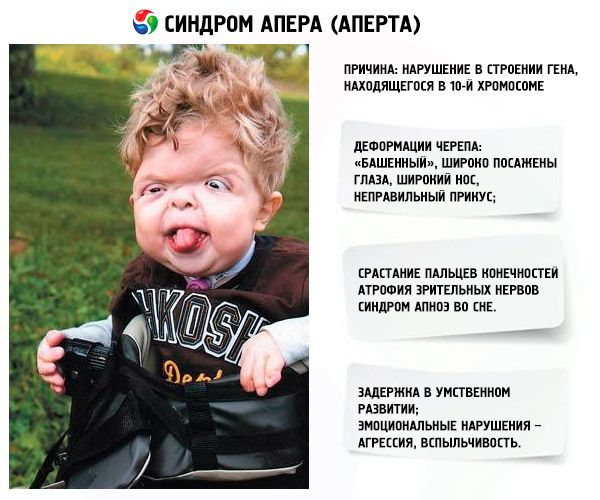
- Czaszka jest zdeformowana - wydłuża się ku górze, przybierając wygląd "wieży", ponadto oczy są szeroko osadzone i lekko wypukłe (gdyż zmniejsza się wielkość oczodołów), nos rozszerza się i kształtuje się nieprawidłowy zgryz (zbyt mocno wystają górne zęby);
- Palce kończyn są całkowicie zrośnięte (głównie palec serdeczny, a także środkowy i wskazujący) i wyglądają jak błona skórna lub całkowite zrośnięcie kości; ponadto mogą urosnąć dodatkowe palce;
- Opóźnienie umysłowe (nie występuje u każdego);
- Nerwy wzrokowe zanikają, w wyniku czego pogarsza się ostrość widzenia (w niektórych przypadkach może dojść do całkowitej utraty wzroku);
- Wzrost ciśnienia śródczaszkowego, który powstaje w wyniku przedwczesnego zamknięcia szwów czaszkowych, objawia się bólami głowy i wymiotami z nudnościami;
- Ponieważ górna szczęka pozostaje słabo rozwinięta, pojawiają się problemy z oddychaniem;
- Zespół bezdechu sennego jest częstym zjawiskiem.
- Objawy emocjonalne – agresja, brak opanowania, duża porywczość.
Komplikacje i konsekwencje
Większość pacjentów ma pewien stopień niedrożności górnych dróg oddechowych w okresie niemowlęcym. Górne drogi oddechowe są słabo drożne z powodu zmniejszonego rozmiaru nosogardła i nozdrzy tylnych, a dolne drogi oddechowe mogą powodować przedwczesną śmierć z powodu nieprawidłowości chrząstki tchawicy.
Diagnostyka Zespół Apera
Aby postawić diagnozę, konieczne jest wykonanie następujących czynności:
- Lekarz powinien przeanalizować skargi pacjenta, a także historię medyczną. Należy dowiedzieć się, czy w rodzinie występowały przypadki podobnej patologii;
- Badanie neurologiczne mające na celu ocenę kształtu czaszki i rozwoju intelektualnego pacjenta (specjalne kwestionariusze i wywiad);
- Badanie dna oka w celu wykrycia obecności objawów wzmożonego ciśnienia śródczaszkowego (obrzęk tarczy nerwu wzrokowego, a także rozmycie jej brzegów);
- Aby ocenić stan czaszki wykonuje się zdjęcie rentgenowskie;
- Komputerowe i magnetyczne obrazowanie głowy w celu zbadania struktury mózgu i czaszki warstwa po warstwie, w celu stwierdzenia obecności objawów przedwczesnego zrośnięcia się szwów czaszkowych, a ponadto wodogłowia (w wyniku wzrostu ciśnienia śródczaszkowego gromadzi się nadmiar płynu mózgowo-rdzeniowego (to właśnie płyn mózgowo-rdzeniowy wspomaga procesy metaboliczne, a także odżywianie mózgu));
- Prześwietlenie stóp i dłoni w celu ustalenia przyczyny zrośnięcia się palców (ma to istotne znaczenie przy planowaniu późniejszej interwencji chirurgicznej);
- Może zostać zalecona konsultacja z genetykiem medycznym i neurochirurgiem.
Testy
Prowadzona jest analiza genetyczna częstych mutacji występujących w genie typu FGFR2.
Diagnostyka różnicowa
Zespół ten należy odróżnić od innych patologii genetycznych, w których obserwuje się kraniosynostozę. Są to choroby takie jak zespoły Pfeiffera, Crouzona, Saethre-Chotzena i Carpentera. W celu wykluczenia tych anomalii stosuje się metody badań genetyki molekularnej.
Z kim się skontaktować?
Leczenie Zespół Apera
Operację uważa się za jedyną skuteczną metodę leczenia zespołu Aperta – pozwala ona skorygować indywidualne defekty fizyczne, a także koryguje upośledzenie umysłowe.
Podczas tej procedury szew koronalny jest zamykany, aby zapobiec możliwemu urazowi mózgu. Najczęstszą techniką jest dystrakcja czaszkowo-twarzowa, która obejmuje stopniową trakcję czaszki. Operacja ortodontyczna i/lub ortognatyczna jest wykonywana w celu usunięcia indywidualnych defektów twarzy.
Dodatkowo pacjenci przechodzą operację korekcji zrostu palców.
Odniesienia
Genetyka medyczna. Przewodnik narodowy. Pod redakcją Gintera EK, Puzyreva VP GEOTAR-Media. 2022