Ekspert medyczny artykułu

Nowe publikacje
Zespół Angelmana u dzieci i dorosłych
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.

Istnieje szereg chorób, w przypadku których wyrażenia takie jak „dbaj o siebie, a nie zachorujesz” brzmią co najmniej śmiesznie. Są to patologie, w których pewne zaburzenia psychiczne i fizyczne są wrodzone w ciele dziecka jeszcze przed jego urodzeniem, ale rodzice nie są za to winni. Takie choroby są spowodowane mutacjami lub nieprawidłowościami w zestawach chromosomowych i nazywane są chromosomalnymi lub genetycznymi. Zespół Angelmana, zespół Downa, zespół Pataua, zespół Edwardsa, zespół Turnera, zespół Pradera-Williego - to tylko część chorób genetycznych z całkiem przyzwoitej listy.
Syndrom szczęśliwego człowieka
Tym razem porozmawiamy o patologii nazwanej na cześć angielskiego pediatry Harry'ego Angelmana, który po raz pierwszy poruszył kwestię tego problemu w 1965 roku, po tym jak poprzedniego dnia spotkał się w swojej praktyce z trójką niezwykłych dzieci, połączonych wspólnymi, osobliwymi objawami. Lekarz nazwał te dzieci dziećmi-lalkami i napisał o nich artykuł, który początkowo nosił tytuł „Dzieci-marionetki”. Sam artykuł i jego tytuł zostały napisane pod wrażeniem obrazu widzianego w jednym z muzeów w Weronie. Obraz przedstawiał śmiejącego się chłopca i nosił tytuł „Chłopiec-marionetka”. Skojarzenie dziecka przedstawionego na obrazie z trójką dzieci, które Angelman kiedyś spotkał w swojej praktyce, skłoniło pediatrę do połączenia dzieci w jedną grupę ze względu na chorobę, na którą cierpiały.
Nie ma nic zaskakującego w tym, że dzieci wymienione w artykule nie zostały zauważone przez innych lekarzy. Wszak na pierwszy rzut oka wydawało się, że mają zupełnie inne choroby, tak różny był ogólny obraz kliniczny choroby w 3 różnych przypadkach. Być może „nowa” patologia chromosomalna zainteresowałaby innych naukowców, ale w tamtym czasie genetyka nie była jeszcze na tyle rozwinięta, aby potwierdzić hipotezę angielskiego lekarza. Dlatego po pewnym zainteresowaniu artykuł został odłożony na później na długi czas.
Kolejna wzmianka o zespole Angelmana, bo tak teraz nazywano artykuł angielskiego pediatry G. Angelmana, pochodzi z początku lat 80. XX wieku. Dopiero w 1987 roku udało się znaleźć przyczynę, dla której niewielka część dzieci rodzi się z takimi odchyleniami, że z zewnątrz wydają się być stale uśmiechnięte i szczęśliwe. W rzeczywistości wcale tak nie jest, a uśmiech to tylko grymas, za którym kryje się nieszczęśliwa dusza człowieka i ból rodziców.
Epidemiologia
Według statystyk mutacja chromosomowa u dziecka może rozwinąć się zarówno na tle podobnych mutacji u rodziców, jak i w ich braku. Nie ma wyraźnej dziedziczności zespołu Angelmana (AS), ale prawdopodobieństwo rozwoju patologii u rodziców z mutacjami chromosomowymi jest dość wysokie.
Ciekawe jest również to, że jeśli w rodzinie jest już dziecko z ZA, istnieje jednoprocentowe prawdopodobieństwo, że drugie dziecko będzie miało to samo zaburzenie, nawet jeśli rodzice są zdrowi.
Nadal nie ma dokładnych statystyk dotyczących liczby chorych na zespół Angelmana. Być może powodem jest różnorodność objawów, które mogą występować w określonym składzie lub nie występować wcale przez długi czas. Przyjmuje się, że częstość występowania choroby wynosi: 1 dziecko na 20 000 noworodków. Ale ta liczba jest bardzo przybliżona.
Przyczyny Zespół Angelmana
Zespół Angelmana to medyczna nazwa patologii chromosomowej, ale nie jest to jedyna taka choroba. Ludzie nazywają tę chorobę zespołem dzieci-lalek, zespołem szczęśliwej kukiełki, zespołem Pietruszki i zespołem śmiejącej się lalki. Ludzie wymyślają najróżniejsze nazwy (czasami nawet obraźliwe dla samych pacjentów i ich rodziców), ale choroba jest chorobą, bez względu na to, jak śmiesznie może wyglądać i bez względu na to, jakie są jej przyczyny.
A przyczynami rozwoju zespołu Angelmana, jak i wielu innych patologii genetycznych, we wszystkich przypadkach są zaburzenia w strukturze jednego z chromosomów lub całego zestawu chromosomów. Ale w naszym przypadku cały problem leży w chromosomie 15, przekazanym przez matkę. To znaczy, że chromosom ojcowski w tym przypadku nie ma żadnych odchyleń, ale żeński ulega pewnym mutacjom.
Zgodnie z rodzajem nieprawidłowości chromosomowej zespół Angelmana jest klasyfikowany jako mutacja chromosomalna. Takie mutacje są uważane za:
- Delecja (brak fragmentu chromosomu zawierającego pewien zestaw genów; jeśli brakuje jednego z genów, mówimy o mikrodelecji), która jest wynikiem dwóch pęknięć i jednego ponownego połączenia, gdy utracony zostaje fragment oryginalnego chromosomu.
- Duplikacja (obecność dodatkowego fragmentu chromosomu, będącego kopią już istniejącego), która w większości przypadków prowadzi do śmierci człowieka, rzadziej do bezpłodności.
- Inwersja (odwrócenie jednego z odcinków chromosomu o 180 stopni, czyli w przeciwnym kierunku, a wówczas geny w nim zawarte znajdują się w odwrotnej kolejności), gdy zerwane końce chromosomu łączą się w kolejności innej niż pierwotna.
- Insercja (jeśli część materiału genetycznego w chromosomie znajduje się w niewłaściwym miejscu),
- translokacja (gdy pewna część chromosomu zostaje przyłączona do innego chromosomu; taka mutacja może być wzajemna bez utraty części).
Otrzymując zmutowany chromosom od niczego niepodejrzewającej matki, dziecko jest skazane na urodzenie się z nieprawidłowościami. Najczęstszą przyczyną zespołu Angelmana jest nadal delecja 15. chromosomu matki, gdy brakuje niewielkiej sekcji. Mniej powszechne mutacje w zespole „śmiejącej się lalki” to:
- translokacja,
- disomia jednoojca (jeśli dziecko otrzymało parę chromosomów od ojca, chromosom matczyny jest nieobecny),
- mutacja genów w DNA, które stanowią zarówno główny materiał budulcowy (genetyczny), jak i instrukcję jego prawidłowego wykorzystania (w szczególności mutacja genu ube3a w chromosomie matczynym).
Obecność jednej z tych mutacji u rodziców jest czynnikiem ryzyka rozwoju zespołu Angelmana u dzieci. Ale nie tylko mutacje chromosomowe, ale także genomiczne (które są związane z ilościową zmianą zestawów chromosomów i są częstsze niż chromosomalne) mogą wywołać rozwój choroby u dziecka. Do powszechnych mutacji genomicznych należy trisomia chromosomowa (jeśli zestaw chromosomów danej osoby ma więcej niż 46 chromosomów).
Aby patologia pojawiła się u dziecka, wcale nie jest konieczne, aby rodzice mieli nieprawidłowości chromosomalne. A jednak istnieje pewien procent pacjentów, których choroba jest dziedziczna.
Patogeneza
Zanurzmy się nieco głębiej w biologię, a dokładniej w genetykę. Informacja genetyczna każdego pojedynczego organizmu ludzkiego zawarta jest w 23 parach chromosomów. Jeden chromosom z pary przekazywany jest dziecku przez ojca, drugi przez matkę. Wszystkie pary chromosomów różnią się kształtem i rozmiarem i niosą pewne informacje. Tak więc 23. para chromosomów (chromosomy X i Y) odpowiada za kształtowanie cech płciowych dziecka (XX - dziewczynka, XY - chłopiec, podczas gdy chromosom Y może być otrzymany przez dziecko tylko od ojca).
W idealnym przypadku dziecko otrzymuje od rodziców 46 chromosomów, które tworzą jego cechy genetyczne, predestynujące je jako jednostkę. Większa liczba chromosomów nazywana jest trisomią i jest uważana za odstępstwo od normy. Na przykład obecność chromosomu 47 w zestawie chromosomów (kariotyp, determinujący cechy gatunkowe i indywidualne) powoduje wystąpienie zespołu Downa.
Jeśli chromosomy zostaną zabarwione specjalnym barwnikiem, to pod mikroskopem można zobaczyć paski o różnych odcieniach wzdłuż każdego z nich. Wewnątrz każdego paska znajduje się ogromna liczba genów. Wszystkie te paski są ponumerowane przez naukowców i mają ustaloną lokalizację. Brak jednego z pasów jest uważany za odstępstwo od normy. W zespole Angelmana bardzo często można zaobserwować brak segmentów chromosomu matczynego w przedziale q11-q13, zlokalizowanym w ramieniu długim, w którym liczba zasad DNA wynosi zaledwie około 4 milionów.
Za główny składnik chromosomu uważa się niewiarygodnie długą cząsteczkę DNA zawierającą tysiące genów i dziesiątki i setki milionów zasad azotowych. Tak więc chromosom 15, odpowiedzialny za rozwój zespołu Angelmana i kilku innych, zawiera 1200 genów i około 100 milionów zasad. Wszelkie zaburzenia w strukturze cząsteczki DNA z pewnością wpłyną na wygląd i rozwój przyszłego dziecka.
Informacje genetyczne zawarte w genach są przekształcane w białko lub RNA. Proces ten nazywa się ekspresją genów. W ten sposób informacje genetyczne otrzymane od rodziców otrzymują zarówno formę, jak i treść, która jest ucieleśniona w ich unikalnym potomku płci żeńskiej lub męskiej.
Istnieje szereg patologii o nieklasycznym sposobie dziedziczenia, w tym zespół Angelmana, w którym geny otrzymane od rodziców jako część sparowanych chromosomów noszą w sobie unikalny ślad rodziców i manifestują się w różny sposób.
Zespół Angelmana jest więc jaskrawym przykładem imprintingu genomowego, w którym ekspresja genów w organizmie dziecka jest bezpośrednio zależna od tego, od którego rodzica otrzymano allele (różne formy jednego genu, otrzymane od ojca i matki, zlokalizowane na identycznych odcinkach sparowanych chromosomów). Oznacza to, że tylko anomalie w chromosomie matczynym prowadzą do rozwoju zespołu, podczas gdy mutacje i zaburzenia strukturalne chromosomu ojcowskiego powodują zupełnie inne patologie.
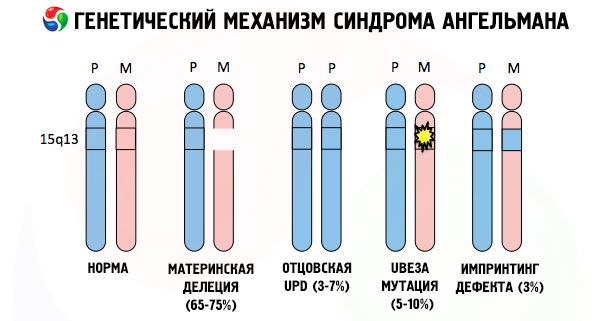
W tej patologii występuje brak pewnych genów w chromosomie matki lub utrata/zmniejszenie aktywności poszczególnych genów (w zdecydowanej większości przypadków jest to gen ube3a, który bierze udział w metabolizmie ubikwityny, białka regulującego degradację innych białek). W rezultacie u dziecka diagnozuje się zaburzenia rozwoju umysłowego i deformacje fizyczne.
Objawy Zespół Angelmana
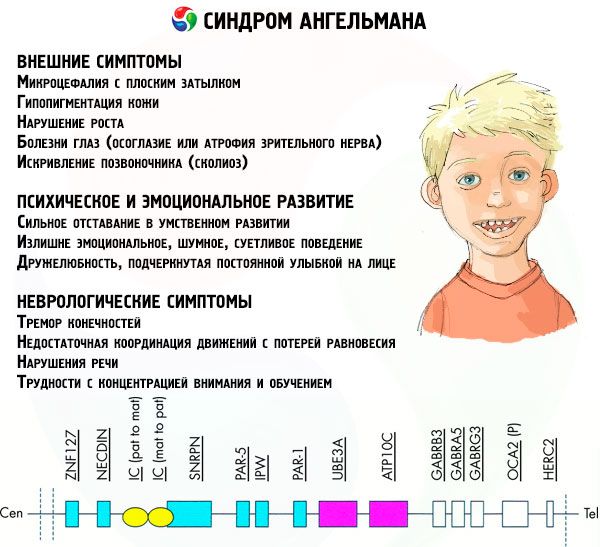
Objawy zespołu Angelmana dotyczą różnych aspektów życia i rozwoju dziecka: fizycznego, neurologicznego, psychicznego. Na tej podstawie można wyróżnić 3 grupy objawów, które wskazują na rozwój tej patologii.
- Objawy zewnętrzne lub fizyczne:
- głowa nieproporcjonalnie mała w porównaniu do ciała i kończyn, które są normalnej wielkości,
- zbyt szerokie usta,
- na twarzy prawie zawsze jest uśmiech (z otwartymi ustami),
- rzadkie zęby,
- wąska górna warga,
- często wystający szeroki język,
- wystająca żuchwa,
- spiczasty podbródek,
- bardzo jasna cera, często owłosienie (albinizm, związany z tym, że organizm nie wytwarza pigmentu melaniny),
- ciemne plamy na jasnej skórze (hipopigmentacja spowodowana niewystarczającą produkcją melaniny)
- objawy fizyczne lub zewnętrzne: choroby oczu, takie jak zez lub zanik nerwu wzrokowego,
- skrzywienie kręgosłupa (skolioza),
- sztywne nogi (podczas chodzenia człowiek nie zgina nóg w kolanach ze względu na małą ruchomość stawów, stąd porównanie do chodu lalki).
- Objawy związane z rozwojem umysłowym i emocjonalnym:
- ciężkie upośledzenie umysłowe,
- nadmiernie emocjonalne, hałaśliwe, wybredne zachowanie,
- częste klaskanie w dłonie,
- wyrażał życzliwość, podkreślaną stałym uśmiechem na twarzy,
- częsty śmiech bez powodu.
- Objawy neurologiczne:
- drżenie kończyn,
- niewystarczająca koordynacja ruchów z utratą równowagi,
- zmniejszone napięcie mięśni,
- różne zaburzenia snu,
- częste napady histerii w dzieciństwie,
- zaburzenia mowy (dziecko zaczyna mówić późno, ma słabe umiejętności komunikacyjne i niewyraźną mowę),
- nadpobudliwość na tle wzmożonej pobudliwości,
- trudności z koncentracją i nauką.
Ale to jest uogólniony obraz choroby. W rzeczywistości obraz kliniczny zespołu Angelmana w dużej mierze zależy od stadium rozwoju choroby i rodzaju mutacji chromosomowej, która spowodowała patologię. Oznacza to, że objawy choroby mogą się znacznie różnić u różnych pacjentów, co przez długi czas nie pozwalało nam odróżnić patologii od innych o podobnym obrazie klinicznym.
Spośród ogólnej liczby objawów można wyróżnić te, które są charakterystyczne dla wszystkich pacjentów bez wyjątku:
- ciężkie upośledzenie umysłowe,
- niewłaściwe zachowanie (nieuzasadniony śmiech, wzmożona pobudliwość, słaba koncentracja, stan euforii),
- niedorozwój umiejętności motorycznych,
- słaba koordynacja ruchów, ataksja chodu (nierównomierne tempo, kołysanie się na boki itp.), drżenie kończyn.
- zaburzenie rozwoju mowy, w którym dominują niewerbalne środki komunikacji.
Wśród objawów, z którymi zmaga się zdecydowana większość pacjentów, można wyróżnić następujące:
- dysproporcja między głową a ciałem spowodowana opóźnionym rozwojem fizycznym,
- u wielu pacjentów kształt czaszki jest taki, że wielkość mózgu pozostaje mniejsza niż u osób zdrowych (małogłowie),
- napady padaczkowe przed ukończeniem 3 roku życia, których siła i częstość stopniowo maleją w późniejszym wieku,
- zniekształcenia parametrów EEG (wahania i wysoka amplituda fal niskiej częstotliwości).
Objawy te są dość powszechne, jednakże 20% pacjentów z zespołem Angelmana ich nie ma.
Jeszcze rzadziej można zdiagnozować takie objawy choroby jak:
- zez ciężki lub łagodny,
- słaba kontrola ruchów języka, co powoduje, że pacjenci często wystawiają język bez powodu,
- trudności z połykaniem i ssaniem, zwłaszcza u małych dzieci,
- zaburzenia pigmentacji skóry i oczu,
- podnoszenie lub zginanie ramion podczas chodzenia,
- hiperrefleksja,
- zaburzenia snu, zwłaszcza w dzieciństwie,
- częste ślinienie się,
- nienasycone pragnienie,
- nadmiernie aktywne ruchy żucia,
- nadwrażliwość na ciepło,
- płaski tył głowy,
- wystająca żuchwa,
- gładkie dłonie.
Dość duży odsetek pacjentów ma problemy z oddawaniem moczu, które słabo kontrolują, upośledzone umiejętności motoryczne, co powoduje trudności w samoopiece i nauce, a także nadwagę. Prawie wszyscy pacjenci przechodzą okres dojrzewania później niż ich zdrowi rówieśnicy.
Dzieci z zespołem Angelmana dobrze odbierają mowę ustną i ją rozumieją, ale nie chcą uczestniczyć w rozmowie, ograniczając swoją mowę do kilkudziesięciu słów niezbędnych w życiu codziennym. Jednak w wieku dorosłym tacy pacjenci wyglądają młodziej niż ich rówieśnicy bez patologii genetycznych.
Wiele objawów zespołu Angelmana jest niestałych, więc obraz kliniczny choroby zmienia się znacząco wraz z wiekiem. Drgawki i napady padaczkowe stają się rzadsze lub całkowicie zanikają, pacjent staje się mniej pobudliwy, a sen się poprawia.
Komplikacje i konsekwencje
Zespół Angelmana to poważna, obecnie praktycznie nieuleczalna patologia chromosomowa, która pozbawia pacjentów możliwości prowadzenia normalnego życia. Jak będzie wyglądało życie dziecka z AS, w dużej mierze zależy od rodzaju nieprawidłowości chromosomowej.
Duplikacja segmentu chromosomu jest w większości przypadków niekompatybilna z życiem. A nawet jeśli tacy pacjenci nie umrą w niemowlęctwie i osiągną dojrzałość płciową, nie mają szans na posiadanie dzieci.
Usunięcie lub brak części genów, które najczęściej występują w zespole Angelmana, jest przeszkodą w nauce chodzenia i mówienia u dziecka. U takich dzieci występuje cięższa postać upośledzenia umysłowego, a napady padaczkowe występują częściej, a ich intensywność jest znacznie większa niż u pacjentów z innymi nieprawidłowościami chromosomowymi.
Jeśli mutacja dotyczy tylko jednego genu, przy należytej uwadze i podejściu dziecko można nauczyć podstaw samoobsługi, komunikacji i interakcji w grupie, choć nadal będzie ono opóźnione w rozwoju względem swoich rówieśników.
Dla dzieci z zespołem Angelmana, które z natury są miłe, najważniejsza jest miłość i uwaga rodziców. Tylko w tym przypadku edukacja dziecka przyniesie owoce, nawet jeśli niewielkie. Oczywiście pacjenci z ZA nie będą mogli uczyć się w zwykłej szkole. Potrzebują specjalnych klas, w których dzieci najpierw nauczą się koncentracji, a następnie stopniowo zostaną im przekazane podstawy wiedzy szkolnej.
Diagnostyka Zespół Angelmana
Zespół Angelmana jest wrodzoną patologią rozwojową. Jednak ze względu na pewne okoliczności często nie można go zdiagnozować w okresie niemowlęcym i wczesnym dzieciństwie. Wynika to z niespecyficzności i słabej ekspresji objawów u niemowląt i dzieci poniżej 3 roku życia. A powszechność choroby w naszym kraju nie jest tak duża, aby lekarze nauczyli się rozpoznawać ją wśród rówieśników.
Zespół Angelmana u niemowląt może objawiać się zmniejszonym napięciem mięśniowym, co objawia się problemami z karmieniem (osłabieniem odruchu ssania i połykania), a później trudnościami w nauce chodzenia (takie dzieci zaczynają chodzić znacznie później). Objawy te są pierwszymi oznakami nieprawidłowości rozwojowych u dziecka, które mogą być związane z nieprawidłowością chromosomową. Tylko analiza genetyczna może potwierdzić to założenie.
Szczególną uwagę zwraca się na dzieci, których rodzice mają różne zaburzenia genomiczne lub chromosomalne. W końcu choroba może nie ujawniać się na początku, a jeśli patologia zostanie wykryta na czas, rozpoczynając intensywną pracę z dzieckiem, można osiągnąć znacznie większy sukces w nauce, spowalniając postęp choroby.
Jeżeli u rodziców występują różnego rodzaju nieprawidłowości chromosomowe, analizę genetyczną przeprowadza się jeszcze przed narodzinami dziecka, ponieważ SA jest jedną z patologii, którą można wykryć już na etapie embrionalnym.
Pobieranie materiału do badań genetycznych może odbywać się na dwa sposoby:
- inwazyjne (obarczone pewnym ryzykiem, gdyż konieczne jest nakłucie macicy w celu pobrania próbki płynu owodniowego),
- nieinwazyjna (analiza DNA dziecka z krwi matki).
Następnie przeprowadza się następujące badania:
- hybrydyzacja fluorescencyjna in situ (metoda FISH) – wiązanie sondy DNA znakowanej specjalnym barwnikiem do badanego DNA, a następnie badanie pod mikroskopem.
- analiza mutacji w genie ube3a i genach imprintowanych,
- Analiza metylacji DNA z wykorzystaniem specjalnych metod stosowanych w genetyce.
Badania genetyczne dostarczają dość dokładnych informacji w przypadku nieprawidłowości chromosomowych, co oznacza, że przyszli rodzice wiedzą z góry, na co się przygotować. Istnieją jednak wyjątki. U pewnej grupy pacjentów, przy obecności wszystkich objawów wskazujących na patologię, wyniki badań pozostają prawidłowe. Oznacza to, że patologię można zidentyfikować tylko poprzez uważną obserwację dziecka od wczesnego dzieciństwa: jak je, kiedy zaczęło chodzić i mówić, czy zgina nogi podczas chodzenia itp.
Oprócz metody FISH, wśród instrumentalnych metod diagnostyki zespołu Angelmana można wyróżnić tomografię komputerową (TK) lub rezonans magnetyczny (MRI), która pozwala określić stan i wielkość mózgu, oraz elektroencefalogram (EEG), który pokazuje, jak pracują poszczególne części mózgu.
Ostateczną diagnozę lekarze zazwyczaj stawiają w wieku 3-7 lat, gdy pacjent ma już większość objawów i widoczna jest dynamika rozwoju choroby.
Jakie testy są potrzebne?
Diagnostyka różnicowa
Zespół Angelmana to patologia genetyczna, która praktycznie nie ma konkretnych objawów. Większość objawów może wskazywać zarówno na AS, jak i inne patologie genetyczne.
Diagnostykę różnicową zespołu Angelmana przeprowadza się w przypadku następujących patologii:
- Zespół Pitta-Hopkinsa (pacjenci charakteryzują się upośledzeniem umysłowym, pogodnym charakterem, uśmiechnięci, mają dość duże i szerokie usta, zauważa się małogłowie). Różnica polega na napadach hiperwentylacji i wstrzymywania oddechu w stanie czuwania.
- Zespół Christiansona (chorzy to osoby upośledzone umysłowo, o pogodnym usposobieniu, niemogące mówić, charakteryzujące się małogłowiem, ataksją, drgawkami i mimowolnymi ruchami mięśni).
- Zespół Mowata-Wilsona (objawy: upośledzenie umysłowe, napady padaczkowe, spiczasty podbródek, otwarte usta, radosny wyraz twarzy, małogłowie). Wyróżnienie: duży rozstaw oczu, oczy skośne do wewnątrz, zaokrąglony czubek nosa, odwrócona małżowina uszna.
- Zespół Kabukiego (charakteryzuje się łagodnym do umiarkowanego upośledzeniem umysłowym, problemami z mową i motoryką, osłabieniem mięśni, napadami padaczkowymi, małogłowiem, długimi przerwami między swędzeniami i zaburzeniami koordynacji). Charakteryzuje się łukowatymi brwiami, wywiniętą boczną częścią dolnej powieki, szeroko rozstawionymi oczami, długimi szparami powiekowymi z długimi, gęstymi rzęsami.
- Zespół Retta (różnicowanie z AS u kobiet). Objawy: opóźniony rozwój mowy, drgawki, małogłowie. Różnica polega na tym, że na twarzy nie ma radosnego wyrazu, występują ataki bezdechu i apraksji, które postępują z czasem.
- Zespół opóźnionego rozwoju umysłowego dziedziczony autosomalnie recesywnie 38 (objawy: wyraźne upośledzenie umysłowe z opóźnieniami w rozwoju motorycznym i mowy, osłabienie mięśni, problemy z karmieniem w niemowlęctwie, impulsywność). Cechą charakterystyczną jest niebieski kolor tęczówki.
- Zespół duplikacji genu MECP 2 (różnicowanie z SA u mężczyzn). Objawy: ciężkie upośledzenie umysłowe, osłabienie mięśni od dzieciństwa, problemy z mówieniem lub brak mówienia, padaczka. Różnice: postępująca miopatia, stale nawracające infekcje.
- Zespół Kleefstry (objawy: problemy z mową i myśleniem, osłabienie mięśni, zaburzenia snu, brak uwagi, otwarte usta, nadpobudliwość, drgawki, ataksja, zaburzenia równowagi). Cechy charakterystyczne: płaska twarz, krótki zadarty nos, szeroko rozstawione oczy, duża wywinięta dolna warga, agresywne wybuchy.
- Zespół Smitha-Magenisa (charakteryzujący się napadami padaczkowymi, problemami ze snem, zaburzeniami rozwoju intelektualnego i motorycznego). Charakterystyczne cechy to szeroka i płaska twarz oraz wystające czoło.
- Zespół Koolena-de Vriesa (łagodne do umiarkowanego upośledzenie umysłowe, osłabienie mięśni, drgawki, przyjacielskość). Cechy charakterystyczne: długa twarz z wysokim czołem, odstające uszy, skośne oczy, duża ruchomość stawów, wrodzone wady serca.
- Zespół Phelana-McDermida (objawy: upośledzenie umysłowe, zaburzenia mowy lub brak mowy). Cechy szczególne: duże dłonie z rozwiniętymi mięśniami, osłabienie mięśni od urodzenia, słabe pocenie się.
Takie patologie jak niedobór adenylobursztynianu, zespół upośledzenia umysłowego dziedziczony autosomalnie recesywnie, zespół duplikacji chromosomu 2q23.1, zespoły haploinsuficjencji genów FOXG1, STXBP1 lub MEF2C i niektóre inne mogą „poszczycić się” objawami podobnymi do zespołu Angelmana.
Zadaniem lekarza jest postawienie trafnej diagnozy, różnicowanie zespołu Angelmana od patologii o podobnych objawach i zalecenie skutecznego leczenia, adekwatnego do rozpoznanego stadium choroby.
Leczenie Zespół Angelmana
Zespół Angelmana jest jedną z tych patologii, dla których medycyna wciąż poszukuje skutecznego leczenia. Leczenie etiologiczne choroby znajduje się w fazie rozwoju różnych metod i środków, z których wiele nie zostało jeszcze przetestowanych na ludziach. Oznacza to, że na razie lekarze muszą ograniczyć się do terapii objawowej, która pomaga w jakiś sposób złagodzić niekorzystną sytuację dzieci i dorosłych z zespołem marionetki, cierpiących na napady padaczkowe, ślinienie, niedociśnienie i zaburzenia snu.
W ten sposób możliwe jest zmniejszenie częstotliwości i siły napadów padaczkowych za pomocą odpowiednio dobranego leku przeciwdrgawkowego. Ale cała trudność polega na tym, że napady u pacjentów z SA różnią się od zwykłych napadów padaczkowych tym, że charakteryzują się kilkoma typami napadów, co oznacza, że stan można złagodzić, podając kilka leków na raz.
Najpopularniejszymi lekami przeciwdrgawkowymi stosowanymi w leczeniu zespołu Angelmana są: kwas walproinowy, topiramat, lamotrygina, lewetyracetam, klonazepam i leki na ich bazie. Rzadziej stosowane są leki na bazie karmazepiny, fenytoiny, fenobarbitalu, etosuksymidu, ponieważ niektóre z nich mogą wywołać paradoksalny efekt polegający na nasileniu i zwiększeniu częstości napadów padaczkowych. Dzieje się tak, jeśli lek jest stosowany w monoterapii.
W leczeniu ślinienia się stosuje się zazwyczaj dwie metody: farmakologiczną (leki hamujące produkcję śliny) i chirurgiczną, która polega na reimplantacji przewodów ślinowych. Jednak w przypadku SA metody te są uważane za nieskuteczne, a kwestia pozostaje otwarta. Rodzice i osoby opiekujące się takimi pacjentami muszą zwrócić szczególną uwagę na tę kwestię, ponieważ sami pacjenci zazwyczaj nie kontrolują ślinienia się, a niektórzy po prostu nie potrafią o siebie zadbać.
Kolejnym problemem jest krótki czas snu. Często dzieci z zespołem Angelmana śpią nie dłużej niż 5 godzin, co negatywnie wpływa na funkcjonowanie całego organizmu. Łatwo pobudliwe, aktywne dzieci, które uwielbiają gry i komunikację (nawet jeśli starają się ograniczyć do metod niewerbalnych) są zauważalnie zmęczone w ciągu dnia. Aby dobrze wypocząć, organizm potrzebuje głębokiego, pełnego snu, ale właśnie w tym tkwi haczyk.
Wydawałoby się, że leki uspokajające (fenotiazyny i atypowe leki przeciwpsychotyczne), które uspokajają układ nerwowy, powinny wystarczyć do poprawy snu u pacjentów pobudliwych. Jednak w przypadku ZZSK stosowanie takich leków jest obarczone wystąpieniem negatywnych skutków. Dlatego lekarze nadal preferują łagodne tabletki nasenne, takie jak melatonina (naturalny lek hormonalny na bazie hormonu snu), którą podaje się pacjentom na godzinę przed pójściem spać w ilości 1 tabletki, oraz difenhydramina. Częstotliwość podawania i dawkowanie ustala lekarz w zależności od stanu i wieku pacjenta.
Czasami pacjenci z zespołem Angelmana mają problemy z trawieniem i stolcem. Możesz poprawić stolec środkami przeczyszczającymi (najlepiej ziołowymi).
Można też podejść do problemu inaczej, jak zrobili to amerykańscy lekarze, opierając się na niektórych metodach leczenia autyzmu, ponieważ wiele objawów charakterystycznych dla ZA jest również charakterystycznych dla autyzmu (impulsywność, ruchy mimowolne, powtarzalne czynności, deficyt uwagi, problemy z komunikacją itp.). Zauważono, że wprowadzenie hormonu sekretyny, który normalizuje trawienie i stolec, ma pozytywny wpływ na uwagę pacjentów, a oksytocyna pomaga poprawić zdolności poznawcze i pamięć dziecka oraz poprawić zachowanie.
Owszem, same hormony nie wystarczą, zwłaszcza jeśli chodzi o dzieci. W zespole Angelmana wskazana jest terapia behawioralna, praca z psychologiem i logopedą (nauka metod komunikacji niewerbalnej i języka migowego). Edukacja takich dzieci powinna opierać się na indywidualnym programie z udziałem specjalnie przeszkolonych nauczycieli, psychologa i rodziców. Niestety nie wszędzie jest to możliwe, a rodziny pozostają same ze swoim problemem.
Ponieważ wielu młodych pacjentów z AS cierpi na niskie napięcie mięśniowe i problemy ze stawami, dużą uwagę przywiązuje się do fizjoterapii. Najczęściej lekarze uciekają się do stosowania aplikacji parafinowych, elektroforezy i terapii magnetycznej.
Aktywny masaż tonizujący i specjalne ćwiczenia terapeutycznego treningu fizycznego pomogą choremu dziecku stanąć na nogach i pewnie chodzić po pewnym czasie. Szczególnie przydatna w tym względzie jest gimnastyka wodna, którą zaleca się SA w chłodnej wodzie. Zwiększa napięcie mięśni i uczy dziecko kontrolowania swojego ciała i koordynacji ruchów.
Leczenie przeciwdrgawkowe
Najbardziej niebezpiecznym objawem zespołu Angelmana są drgawki podobne do padaczki. Objaw ten występuje u 80% pacjentów, co oznacza, że wszyscy muszą otrzymać skuteczne leczenie przeciwdrgawkowe.
Leczenie napadów padaczkowych odbywa się za pomocą witamin i leków przeciwdrgawkowych. W zespole Angelmana, któremu towarzyszy zespół drgawkowy, przydatne będą witaminy z grupy B, a także witaminy C, D i E. Jednak samodzielne przepisywanie terapii witaminowej w tym przypadku jest bardzo niebezpieczne, ponieważ niekontrolowane przyjmowanie witamin może zmniejszyć skuteczność leków przeciwpadaczkowych i wywołać nowe, cięższe i dłuższe napady.
Dobór leków przeciwdrgawkowych i przepisanie ich skutecznej dawki powinien również zostać dokonane przez lekarza specjalistę. On również zdecyduje, czy wystarczy jeden lek, czy pacjent będzie musiał przyjmować 2 lub więcej leków przez dłuższy czas.
Większości pacjentów lekarze przepisują leki zawierające kwas walproinowy (kwas walproinowy, Depakine, Convulex, Valparin itp.), które zapobiegają napadom padaczkowym oraz poprawiają nastrój i stan psychiczny pacjentów.
Kwas walproinowy jest dostępny w postaci tabletek, syropu i roztworów do wstrzykiwań. Najpopularniejszym lekiem jest lek o przedłużonym uwalnianiu „Depakine” w tabletkach i jako roztwór do podawania dożylnego. Dawkowanie leku ustala lekarz indywidualnie w zależności od masy ciała, wieku i stanu pacjenta.
Lek przyjmuje się podczas posiłków 2 do 3 razy dziennie. Średnia dawka dobowa wynosi 20-30 mg na 1 kilogram masy ciała pacjenta, maksymalna dawka to 50 mg/kg na dobę.
Przeciwwskazania do stosowania. Nie stosować w przypadku dysfunkcji wątroby i trzustki, skazy krwotocznej, zapalenia wątroby, porfirii i nadwrażliwości na lek.
Do skutków ubocznych zalicza się drżenie rąk, zaburzenia trawienia i stolca oraz wahania masy ciała.
„Topiramat” jest również lekiem pierwszego wyboru w przypadku SA. Jest produkowany w formie tabletek i jest stosowany zarówno jako część monoterapii, jak i w połączeniu z innymi lekami.
Sposób podawania i dawkowanie. Tabletki należy przyjmować doustnie, niezależnie od przyjmowania pokarmu. Początkowa dawka dobowa dla dorosłych wynosi 25-50 mg, dla dzieci - 0,5-1 mg/kg. Co tydzień dawkę zwiększa się zgodnie z zaleceniami lekarza.
Leku nie należy stosować w czasie ciąży i karmienia piersią, a także w przypadku nadwrażliwości na jego składniki. Lek ma wiele różnych działań niepożądanych.
Leki, które lekarz może przepisać na zespół Angelmana: Klomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra itp.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Medycyna tradycyjna i homeopatia
Medycyna tradycyjna, na przykład preparaty homeopatyczne, jest oczywiście stosunkowo bezpieczna, ale skuteczność takiego leczenia zespołu Angelmana można uznać za kontrowersyjną.
Chociaż leczenie ludowe nadal może pomóc w niektórych sprawach. Mówimy o zatrzymaniu napadów padaczkowych. W tym względzie leczenie ziołowe może być dość skuteczne.
Dobry efekt daje zbiór leczniczy na bazie piwonii, lukrecji i rzęsy wodnej (składniki przyjmuje się w równych ilościach). Zioła należy zmielić na mąkę. Po 2 tygodniach od rozpoczęcia przyjmowania można zauważyć znaczny spadek częstotliwości napadów.
Na skurcze pomocny jest również wywar z lawendy (1 łyżeczka na szklankę wrzątku). Mieszankę gotuje się przez 5 minut i zaparza przez pół godziny. Lek przyjmuje się na noc przez 14 dni.
Napar wodny (lub alkoholowy) z ziela serdecznika jest skuteczny w leczeniu napadów padaczkowych.
Spośród preparatów homeopatycznych zapobiegających napadom padaczkowym w zespole Angelmana można stosować leki na bazie rumianku i serdecznika, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Należy jednak wziąć pod uwagę, że tylko lekarz homeopata może przepisać skuteczne i bezpieczne dawki leków w każdym konkretnym przypadku.
Zapobieganie
Jak czytelnik prawdopodobnie już zrozumiał, medycyna nie jest jeszcze w stanie zapobiec mutacjom genów i innym nieprawidłowościom chromosomowym, ani też naprawić sytuacji. Może się to zdarzyć każdemu, ponieważ dzieci z zespołem Angelmana rodzą się zdrowym rodzicom, a genetyka, która jest obecnie jedną z najmniej zbadanych dziedzin medycyny, nie potrafi jeszcze tego wyjaśnić.
Jedyne, co można zrobić, to odpowiedzialnie podejść do planowania ciąży, zarejestrować się i wykonać badania w odpowiednim czasie. Ale znowu, takie działanie będzie bardziej edukacyjne niż profilaktyczne, jak każde badanie. Ale młodzi rodzice będą wiedzieć z góry, na co się przygotować, a w przypadku pozytywnej odpowiedzi podejmą decyzję, czy mogą wziąć na siebie taką odpowiedzialność, jak wychowanie chorego dziecka.
Prognoza
Rokowanie w zespole Angelmana zależy od charakteru nieprawidłowości chromosomowej i terminowości jej wykrycia. Najbardziej dotknięte są dzieci, których chromosom 15 zawiera „luki” w genach (delecje). Prawdopodobieństwo, że tacy pacjenci będą chodzić i mówić, jest niezwykle niskie. Inne przypadki można skorygować dzięki ostrożnemu podejściu i miłości do dziecka.
Niestety tacy pacjenci nie będą mogli stać się pełnoprawnymi członkami społeczeństwa, mimo że są dalecy od głupoty, rozumieją mowę i jej znaczenie. Będą jednak mieli problemy z komunikacją do końca życia. Pacjentów można uczyć języka migowego od dzieciństwa, ale nie można ich zmusić do komunikowania się za pomocą słów. Słownictwo pacjentów „mówiących” ogranicza się do minimum słów używanych w życiu codziennym (5-15 słów).
Jeśli chodzi o oczekiwaną długość życia i ogólny stan zdrowia pacjentów z zespołem Angelmana, liczby te oscylują wokół wartości średnich. W wieku dorosłym pacjenci najczęściej borykają się z problemami zdrowotnymi, takimi jak skolioza i otyłość, które przy odpowiednim podejściu do leczenia nie zagrażają życiu.