Ekspert medyczny artykułu

Nowe publikacje
Zespół Treachera Collinsa
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.

Zaburzenia wewnątrzmaciczne w procesach rozwoju kości są przyczyną poważnych deformacji twarzoczaszki, a jedną z odmian tej patologii jest zespół Treachera Collinsa (TCS) lub dysostoza żuchwowo-powięziowa, czyli szczękowo-twarzowa.
Kod choroby według ICD 10: klasa XVII (wady wrodzone, deformacje i zaburzenia chromosomowe), Q75.4 - dyzostoza żuchwowo-twarzowa.
Przyczyny Zespół Treachera Collinsa
Zespół ten został nazwany na cześć wybitnego brytyjskiego okulisty Edwarda Treachera Collinsa, który opisał główne cechy tej patologii ponad sto lat temu. Jednak europejscy lekarze częściej nazywają ten typ anomalii kości twarzy i szczęki chorobą lub zespołem Franceschettiego - na podstawie szeroko zakrojonych badań szwajcarskiego okulisty Adolfa Franceschettiego, który wprowadził termin „dyzostoza żuchwowo-powięziowa” w połowie ubiegłego wieku. W kręgach medycznych stosuje się również nazwę zespół Franceschettiego-Collinsa.
Zespół Treachera Collinsa jest spowodowany mutacjami w genie TCOF1 (w locus chromosomu 5q31.3-33.3), który koduje fosfoproteinę jąderkową odpowiedzialną za formowanie części twarzowo-czaszkowej zarodka ludzkiego. W wyniku przedwczesnego zmniejszenia ilości tego białka dochodzi do zaburzenia biogenezy i funkcji rRNA. Według genetyków z programu badawczego Human Genome procesy te prowadzą do zmniejszenia proliferacji komórek embrionalnych grzebienia nerwowego – grzbietu wzdłuż bruzdy nerwowej, który w trakcie rozwoju embrionalnego zamyka się w cewę nerwową.
Powstawanie tkanek twarzy następuje w wyniku transformacji i różnicowania komórek górnej (głowowej) części grzebienia nerwowego, które migrują wzdłuż cewy nerwowej do obszaru pierwszego i drugiego łuku skrzelowego zarodka. A niedobór tych komórek powoduje deformacje twarzoczaszki. Krytyczny okres występowania anomalii przypada na okres od 18 do 28 dni po zapłodnieniu. Po zakończeniu migracji komórek grzebienia nerwowego (w czwartym tygodniu ciąży) powstają prawie wszystkie luźne tkanki mezenchymalne w okolicy twarzy, które później (od 5 do 8 tygodni) różnicują się w tkanki szkieletowe i łączne wszystkich części twarzy, szyi, krtani, ucha (w tym ucha wewnętrznego) i przyszłych zębów.
Patogeneza
Patogeneza zespołu Treachera Collinsa jest często rodzinna, a anomalia dziedziczona jest w sposób autosomalny dominujący, chociaż zdarzają się przypadki autosomalnego recesywnego przekazywania defektu (z mutacjami w innych genach, w szczególności POLR1C i POLR1D). Najbardziej nieprzewidywalną rzeczą w przypadku dysostozy szczękowo-twarzowej jest to, że mutacja jest dziedziczona przez dzieci tylko w 40-48% przypadków. Oznacza to, że u 52-60% pacjentów przyczyny zespołu Treachera Collinsa nie są związane z obecnością anomalii w rodzinie i uważa się, że patologia występuje w wyniku sporadycznych mutacji genów de novo. Najprawdopodobniej nowe mutacje są konsekwencją teratogennych efektów na płód w czasie ciąży.
Wśród przyczyn teratogennych tego zespołu eksperci wymieniają duże dawki etanolu (alkoholu etylowego), promieniowanie, dym papierosowy, cytomegawirus i toksoplazmę, a także herbicydy na bazie glifosatu (Roundal, Glyfor, Tornado itp.). A lista czynników jatrogennych obejmuje leki na trądzik i łojotok z kwasem 13-cis-retinowym (Isotretinoin, Accutane); lek przeciwdrgawkowy Phenytoin (Dilantin, Epanutin); leki psychotropowe Diazepam, Valium, Relanium, Seduxen.
Objawy Zespół Treachera Collinsa
W większości przypadków objawy kliniczne dysostozy żuchwowo-powięziowej i stopień ich ekspresji zależą od cech manifestacji mutacji genowych. A pierwsze objawy tej anomalii w większości przypadków są widoczne u dziecka zaraz po urodzeniu: twarz z zespołem Treachera Collinsa ma charakterystyczny wygląd. Ponadto anomalie morfologiczne są zwykle obustronne i symetryczne.
Najbardziej oczywiste objawy zespołu Treachera Collinsa to:
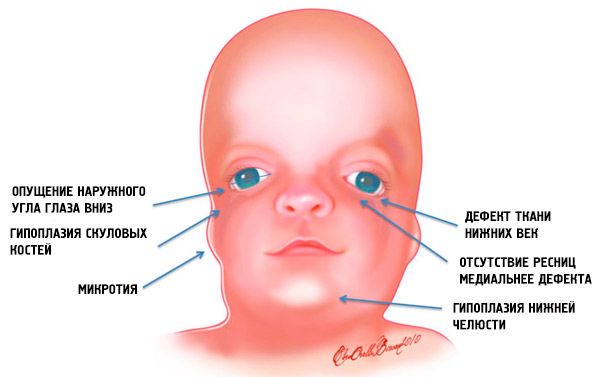
- niedorozwój (hipoplazja) kości twarzowej czaszki: kości jarzmowej, wyrostków jarzmowych kości czołowej, blaszek skrzydłowych bocznych, zatok przynosowych, żuchwy i wypustek nasad kości (kłykci);
- niedorozwój kości żuchwy (mikrognacja) i bardziej rozwarty niż zwykle kąt żuchwy;
- nos ma normalną wielkość, ale wydaje się duży z powodu niedorozwoju łuków brwiowych i niedorozwoju lub braku łuków jarzmowych w okolicy skroniowej;
- szpary powiekowe skierowane są ku dołowi, tzn. kształt oczu jest nieprawidłowy, a zewnętrzne kąciki opadają ku dołowi;
- defekty powiek dolnych (coloboma) i częściowy brak rzęs na nich;
- nieregularny kształt małżowin usznych, z szerokim zakresem odchyleń, w tym ich położenie w kąciku żuchwy, brak płatków, ślepe przetoki między skrawkiem ucha a kącikiem ust itp.;
- zwężenie lub zamknięcie (atrezja) przewodu słuchowego zewnętrznego oraz anomalie kosteczek słuchowych ucha środkowego;
- brak lub niedorozwój ślinianek przyusznych;
- hipoplazja gardła (zwężenie gardła i dróg oddechowych);
- brak zrośnięcia się podniebienia twardego (rozszczep podniebienia), a także brak, skrócenie lub unieruchomienie podniebienia miękkiego.
Takie anomalie anatomiczne we wszystkich przypadkach mają powikłania. Są to zaburzenia czynnościowe słuchu w postaci niedosłuchu przewodzeniowego lub całkowitej głuchoty; upośledzenie wzroku spowodowane nieprawidłowym uformowaniem gałek ocznych; wady podniebienia powodujące trudności z karmieniem i połykaniem. Istnieją zaburzenia zgryzu zębowego (malokluzja) związane z wadami szczęki, co z kolei powoduje problemy z żuciem i artykulacją. Patologie podniebienia miękkiego wyjaśniają głos nosowy.
Komplikacje i konsekwencje
Konsekwencją anomalii szczękowo-twarzowych w zespole Treachera-Collinsa jest to, że przy urodzeniu dziecko ma prawidłowe zdolności intelektualne, ale z powodu wad słuchu i innych zaburzeń obserwuje się wtórne upośledzenie umysłowe.
Ponadto dzieci z takimi defektami odczuwają dotkliwie swoją niższość i cierpią, co negatywnie odbija się na ich układzie nerwowym i psychice.
Diagnostyka Zespół Treachera Collinsa
Postnatalne rozpoznanie zespołu Treachera Collinsa opiera się zasadniczo na objawach klinicznych. Dyzostozę czaszkowo-twarzową łatwo zidentyfikować, gdy zespół jest w pełni ekspresyjny, ale gdy występują minimalnie wyrażone objawy patologii, mogą pojawić się problemy z ustaleniem prawidłowej diagnozy.
W tym przypadku należy zwrócić szczególną uwagę na ocenę wszystkich funkcji związanych z anomaliami, zwłaszcza tych wpływających na oddychanie (ze względu na ryzyko bezdechu sennego). Należy również ocenić i monitorować skuteczność karmienia i wysycenie hemoglobiny tlenem.
Później, w 5–6 dniu po porodzie, konieczne będzie określenie stopnia uszkodzenia słuchu za pomocą badań audiologicznych, które należy przeprowadzić w szpitalu położniczym.
Zalecane jest przeprowadzenie badania, w trakcie którego przeprowadza się diagnostykę instrumentalną, obejmującą: prześwietlenie rentgenowskie czaszki w celu wykrycia dysmorfii twarzoczaszki, pantomografię (panoramiczne zdjęcie rentgenowskie struktur kostnych czaszki twarzowej), pełną tomografię komputerową czaszki w różnych projekcjach, tomografię komputerową lub rezonans magnetyczny mózgu w celu określenia stanu przewodu słuchowego wewnętrznego.
Najwcześniejsze – prenatalne – rozpoznanie anomalii szczękowo-twarzowych przy występowaniu zespołu Treachera-Collinsa w wywiadzie rodzinnym możliwe jest na podstawie biopsji kosmówki wykonanej w 10-11 tygodniu ciąży (zabieg ten grozi poronieniem i zakażeniem macicy).
Od członków rodziny pobiera się również krew; w 16-17 tygodniu ciąży wykonuje się analizę płynu owodniowego (amniopunkcja przezbrzuszna), a w 18-20 tygodniu ciąży wykonuje się fetoskopię i pobiera krew z naczyń płodowych łożyska.
Najczęściej jednak badanie ultrasonograficzne stosuje się w prenatalnej diagnostyce tego zespołu u płodu (w 20–24 tygodniu ciąży).
Jakie testy są potrzebne?
Diagnostyka różnicowa
Te same metody stosują specjaliści w przypadku konieczności przeprowadzenia diagnostyki różnicowej w celu rozpoznania łagodnego zespołu Treachera Collinsa i odróżnienia go od innych wrodzonych anomalii kości twarzoczaszki, w szczególności: zespołów Aperta, Crouzona, Nagera, Petersa-Hewelsa, Hellermanna-Stepha, a także mikrosomii połowiczej twarzy (zespołu Goldenhara), hiperteloryzmu, przedwczesnego zrastania się szwów czaszkowych (kraniosynostozy) lub zaburzonego zrastania się kości twarzoczaszki (kraniosynostozy).
Leczenie Zespół Treachera Collinsa
Jak we wszystkich przypadkach genetycznie uwarunkowanych wad wrodzonych, leczenie ciężkich postaci zespołu Treachera Collinsa jest wyłącznie paliatywne, ponieważ po prostu nie ma metod terapeutycznych na takie patologie. Spektrum i stopień deformacji w tym zespole są rozległe, a zatem charakter i intensywność interwencji medycznej również mają wiele opcji.
Aparaty słuchowe służą do korekcji i poprawy słuchu, natomiast sesje terapii mowy służą do poprawy mowy.
W przypadku poważnych przypadków zwężenia dróg oddechowych (wykonuje się tracheostomię) i krtani (w celu karmienia wykonuje się gastrostomię) interwencje chirurgiczne są wymagane we wczesnym wieku. Może być również wymagana chirurgiczna korekta podniebienia.
Operacje wydłużania żuchwy wykonuje się w wieku 2-3 lat lub później. Rekonstrukcja tkanek miękkich obejmuje korekcję coloboma powieki dolnej i chirurgię plastyczną małżowiny usznej.
Zapobieganie
Prognoza
Jakie jest rokowanie w przypadku tej patologii? Zależy ono od stopnia deformacji i nasilenia objawów. Zespół Treachera Collinsa to diagnoza na całe życie.
 [ 25 ]
[ 25 ]