Ekspert medyczny artykułu

Nowe publikacje
Alkaptonuria jest wrodzoną nieprawidłowością enzymatyczną
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Jedno z bardzo rzadkich zaburzeń metabolicznych, alkaptonuria, to wrodzone anomalie metabolizmu aminokwasu tyrozyny.
Zespół ten może być również nazywany niedoborem oksydazy homogentyzynianowej, homogentynurią, dziedziczną ochronozą lub chorobą czarnego moczu.[ 1 ]
Epidemiologia
Według statystyk, na 1 milion osób przypada nie więcej niż dziewięć przypadków alkaptonurii. A w większości krajów europejskich jeden przypadek przypada na 100-250 tysięcy żywych urodzeń.
Wśród krajów europejskich wyjątkiem jest Słowacja (zwłaszcza stosunkowo niewielki region północno-zachodni), gdzie częstość występowania alkaptonurii wynosi jeden przypadek na 19 000 noworodków. Wynika to najprawdopodobniej z faktu, że wśród mieszkających tam słowackich rodzin romskich poziom chowu wsobnego (małżeństw między kuzynami) jest najwyższy w Europie: 10-14%. [ 2 ]
Przyczyny alkaptonuria
Dokładne przyczyny alkaptonurii, jako wrodzonego zaburzenia katabolizmu (rozpadu metabolicznego) aromatycznego (homocyklicznego) α-aminokwasu tyrozyny, zostały ustalone: ten typ zaburzenia metabolicznego jest konsekwencją homozygotycznych lub złożonych heterozygotycznych mutacji jednego z tysięcy genów na chromosomie 3, a dokładniej genu HGD w locus 3q21-q23 na długim ramieniu chromosomu. Gen ten koduje sekwencje nukleotydowe enzymu wątrobowego homogentyzynian-1,2-dioksygenazy [ 3 ] (zwanej również oksydazą kwasu homogentyzynowego lub oksydazą homogentyzynianową) - metaloproteiny zawierającej żelazo, niezbędnej do jednego z etapów rozkładu tyrozyny w organizmie. [ 4 ], [ 5 ]
Alkaptonuria jest zatem defektem enzymu homogentyzynian-1,2-dioksygenazy, a dokładniej wynikiem jego uwarunkowanego genetycznie niedoboru lub całkowitego braku. [ 6 ]
Będąc wrodzonym niedoborem enzymu, alkaptonuria dziedziczona jest jako cecha autosomalna recesywna. Oznacza to, że aby alkaptonuria wystąpiła u dzieci, oboje rodzice muszą mieć zmodyfikowany gen kodujący enzym, ponieważ każdy z nich przekazuje dziecku tylko jedną kopię genu spośród dwóch dostępnych.
Według najnowszych danych istnieje ponad dwieście wariantów modyfikacji genu HGD, przy czym najczęściej obserwuje się mutacje typu missense, translokacje i splicing.
Czynniki ryzyka
Jedynym czynnikiem ryzyka rozwoju tej wrodzonej enzymopatii jest jej obecność w historii rodzinnej oraz odziedziczenie dwóch zmodyfikowanych kopii genu HGD, jeśli rodzice nie wykazują alkaptonurii (ryzyko przekazania anomalii wynosi 25%) lub jedno z rodziców ma to zaburzenie. [ 7 ]
Patogeneza
Tyrozyna odgrywa kluczową rolę w syntezie białek, produkcji chromoprotein – pigmentu skóry – melaniny, a także hormonów tarczycy i neuroprzekaźników katecholaminowych.
Mechanizm regulacji ilości tyrozyny w komórkach jest bardzo złożony, a organizm normalizuje jej nadmiar poprzez jej rozkład. Proces katabolizmu tyrozyny, podobnie jak wszystkich aminokwasów aromatycznych, jest wieloetapowy i przebiega w kilku etapach. Każdy etap metabolicznego rozkładu tyrozyny zachodzi przy udziale określonego enzymu i tworzeniu związku pośredniego.
Tak więc najpierw aminokwas rozkłada się do para-hydroksyfenylopirogronianu, który przekształca się w alkapton – kwas 2,5-dihydroksyfenylooctowy lub homogentyzynowy. Następnie alkapton powinien zostać przekształcony w kwas maleoctowy, ale tak się nie dzieje. [ 8 ]
A patogeneza alkaptonurii polega na ustaniu reakcji biochemicznych katabolizmu tyrozyny na etapie powstawania kwasu homogentyzynowego: po prostu nie ma enzymu potrzebnego do jego rozkładu – oksydazy homogentyzynowej.
Kwas homogentyzynowy nie jest wykorzystywany przez organizm i może gromadzić się wraz z wydalaniem przez nerki. Ponadto jest utleniany do benzochinooctanu (kwasu benzochinonooctowego), który wiążąc się z cząsteczkami tkanek i płynów ustrojowych, tworzy związki biopolimerowe w kolorze melaniny.
Gromadzenie się tych pośrednich produktów w tkance prowadzi do zaburzenia struktury kolagenowej tkanki chrzęstnej, co powoduje zmniejszenie jej elastyczności – z pojawieniem się wielu objawów klinicznych alkaptonurii i rozwojem powikłań.
Objawy alkaptonuria
Alkaptonuria u noworodków i niemowląt charakteryzuje się ciemnieniem moczu. Po wystawieniu na działanie powietrza mocz na pieluchach, pieluszkach i bieliźnie staje się ciemnobrązowy; jest to spowodowane gromadzeniem się i uwalnianiem kwasu homogentyzynowego, który utlenia się do benzochinooctanu. [ 9 ]
W przypadku braku innych objawów alkaptonuria u małych dzieci często nie jest rozpoznawana w odpowiednim czasie, ponieważ mocz może ciemnieć po kilku godzinach oddawania moczu. Według niektórych danych, tylko jedna piąta dzieci poniżej 12 miesiąca życia, które urodziły się z niedoborem tego enzymu, jest identyfikowana w warunkach klinicznych. Dlatego bardzo ważne jest, aby rodzice zwracali uwagę na opiekę nad swoimi niemowlętami.
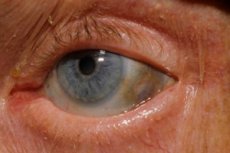
Ponadto do wczesnych objawów zalicza się pigmentację (niebiesko-szarą barwę) twardówki oka oraz chrząstek uszu i nosa, co często nazywa się ochronozą.[ 10 ]
Z czasem pojawiają się inne objawy:
- silna pigmentacja skóry na kościach policzkowych, pachach i narządach płciowych;
- plamienie odzieży w kontakcie ze spoconymi częściami ciała;
- ataki ogólnego osłabienia;
- chrypliwy głos.
Należy pamiętać, że alkaptonuria i ochronoza, jak wspomniano powyżej, są synonimami tego samego zaburzenia katabolizmu tyrozyny.
Choroba syropu klonowego i alkaptonuria. Wrodzona choroba syropu klonowego lub leucynoza jest również zaburzeniem metabolicznym, ma ten sam wzór dziedziczenia, a nawet mutacje występują na tym samym chromosomie, ale wpływają na gen kodujący kompleks enzymatyczny rozgałęzionej dehydrogenazy α-ketokwasu. Z tego powodu organizm nie może rozkładać niektórych składników białek, w szczególności aminokwasów leucyny, izoleucyny i waliny. W przypadku tej choroby mocz (i woskowina) mają słodki zapach; ponadto obraz kliniczny tego typu kwasicy organicznej obejmuje hipopigmentację, wahania ciśnienia krwi, drgawki, wymioty i biegunkę, spadek poziomu glukozy we krwi, kwasicę ketonową, halucynacje itp. Śmiertelność u dzieci jest dość wysoka; u dorosłych bez leczenia może wystąpić śpiączka i śmierć z powodu obrzęku mózgu.
Albinizm i alkaptonuria są „zjednoczone” tylko przez tyrozynę. Albinizm, w tym oczno-skórny, jest spowodowany mutacjami genetycznymi, które wpływają na produkcję pigmentu melaniny. Wrodzone zmiany są obserwowane w genie TYR na chromosomie 11 (11q14.3), który koduje tyrozynazę, zawierający miedź enzym melanosomu niezbędny do tworzenia pigmentu skóry na podstawie produktów metabolizmu tyrozyny. Ta choroba jest znacznie bardziej powszechna niż alkaptonuria.
Komplikacje i konsekwencje
W wyniku działania pośrednich metabolitów tyrozyny – kwasu homogentyzynowego i benzochinonooctowego – dochodzi do następstw i powikłań alkaptonurii, które objawiają się odkładaniem się reaktywnych polimerów barwnikowych, niszczeniem włókienek kolagenowych i pogorszeniem stanu chrząstki (ze spadkiem jej odporności na obciążenia mechaniczne).
Z biegiem lat, w wieku dorosłym, rozwija się zwyrodnienie stawów i choroba zwyrodnieniowa dużych stawów (biodrowych, krzyżowo-biodrowych i kolanowych); przestrzenie międzykręgowe zwężają się (szczególnie w kręgosłupie lędźwiowym i piersiowym) – z wapnieniem i tworzeniem osteofitów; gęstość tkanki podchrzęstnych blaszek kostnych zmniejsza się, a znajdujące się pod nimi kości mogą ulegać patologicznej przebudowie z tworzeniem narośli i deformacji. [ 11 ]
W wyniku tego samego zwapnienia mogą wystąpić uszkodzenia zastawek serca (aorty i mitralnej) oraz tętnic wieńcowych – z objawami choroby niedokrwiennej serca, a także tworzenie się kamieni w nerkach i gruczole krokowym. [ 12 ], [ 13 ]
Diagnostyka alkaptonuria
Zazwyczaj rozpoznanie wrodzonych zaburzeń metabolicznych opiera się na badaniu płynów biologicznych organizmu.
Na podstawie jakich badań i reakcji można zdiagnozować alkaptonurię? Badania moczu są wymagane do wykrycia kwasu homogentyzynowego i określenia jego poziomu (normalny – 20-30 mg na dobę, podwyższony – 3-8 g). Próbkę moczu bada się chromatografią gazową lub spektrometrią masową, stosując chromatografię cieczową; możliwe jest badanie przesiewowe na obecność chlorku żelaza w moczu. [ 14 ]
Istnieje również metoda szybkiej diagnostyki – oznaczanie alkaptonów w zaschniętych plamach moczu na papierze (na podstawie intensywności barwy).
W celu ustalenia rozpoznania stosuje się diagnostykę instrumentalną (radiografię), która polega na określeniu u chorych radiologicznych objawów choroby zwyrodnieniowej stawów i innych patologii stawów.
Rozpoznanie potwierdza się metodami genetyki molekularnej stosowanymi w diagnostyce chorób dziedzicznych, takimi jak badania genetyczne i sekwencjonowanie DNA. [ 15 ]
Diagnostyka różnicowa
Diagnostyka różnicowa obejmuje hemochromatozę i ostrą niewydolność wątroby noworodków, melaninurię, ostrą porfirię przerywaną, limfohistiocytozę hemofagocytarną, pierwotną patologię mitochondrialną, reumatoidalne zapalenie stawów, zesztywniające zapalenie stawów kręgosłupa.
Z kim się skontaktować?
Leczenie alkaptonuria
Głównym leczeniem alkaptonurii jest doustne podawanie dużych dawek (co najmniej 1000 mg na dobę) kwasu askorbinowego. U dzieci zwiększa to wydalanie kwasu homogentyzynowego z moczem, a u dorosłych zmniejsza zawartość w moczu jego pochodnej, kwasu benzochinonooctowego, i spowalnia jego wiązanie ze strukturami tkanki łącznej stawów i kolagenem. [ 16 ]
Kliniki zachodnioeuropejskie testują lek Nitisinone (Orfalin), lek z grupy metabolitów, który hamuje drugi etap katabolizmu tyrozyny: przekształcanie para-hydroksyfenylopirogronianu w kwas homogentyzynowy. Jednak stosowanie tego środka farmakologicznego prowadzi do kumulacji tyrozyny i może powodować poważne skutki uboczne, w tym zmętnienie rogówki i światłowstręt, krwawienia z nosa i żołądka, niewydolność wątroby, zmiany we krwi itp. Niemniej jednak w Stanach Zjednoczonych Nitisinone jest zatwierdzony przez FDA do leczenia tyrozynemii typu I. [17 ], [ 18 ]
W związku z tym w przypadku problemów ze stawami wywołanych alkaptonurią stosuje się leczenie fizjoterapeutyczne – terapię ruchową mającą na celu zwiększenie siły mięśni i poprawę ruchomości stawów, balneoterapię i peloidoterapię mającą na celu ograniczenie bólu.
Mimo że tyrozyna nie jest dostarczana wyłącznie z pożywieniem, ale również jest wytwarzana w organizmie, pacjentom z alkaptonurią zaleca się stosowanie diety niskobiałkowej i ograniczenie spożycia produktów bogatych w tyrozynę, przede wszystkim wołowiny i wieprzowiny, produktów mlecznych (zwłaszcza serów), roślin strączkowych, orzechów i nasion.
Zapobieganie
Zapobieganie mutacjom genetycznym jest niemożliwe, ale aby zapobiec narodzinom dzieci z dużym ryzykiem wystąpienia wad wrodzonych, konieczne jest skorzystanie z poradnictwa genetycznego przed planowaną ciążą u par, w których rodzinie występują choroby dziedziczne. [ 19 ]
Prognoza
Śmiertelne skutki alkaptonurii są bardzo rzadkie, a śmierć może być spowodowana poważnymi powikłaniami obejmującymi serce i nerki. Tak więc ogólna oczekiwana długość życia osób z alkaptonurią jest dobra.
Jednak jakość życia ulega pogorszeniu z powodu silnego bólu stawów lub kręgosłupa, któremu towarzyszy znaczne ograniczenie ruchomości, często postępujące.