Ekspert medyczny artykułu

Nowe publikacje
Xeroderma pigmentosum
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
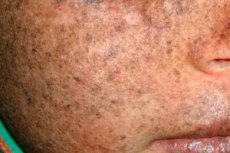
Xeroderma pigmentosum to autosomalne recesywne zaburzenie genetyczne naprawy DNA. Choroba jest spowodowana mutacjami w genach, które biorą udział w naprawie DNA uszkodzonego przez promieniowanie ultrafioletowe i inne substancje toksyczne.
Najczęściej choroba ta dotyka dzieci, które są również nazywane „dziećmi nocy”. Częstymi powikłaniami choroby są rak podstawnokomórkowy i inne nowotwory złośliwe skóry, przerzutowy czerniak złośliwy i rak płaskonabłonkowy.
 [ 1 ]
[ 1 ]
Przyczyny xeroderma pigmentosum
Patogeneza
Niedobór enzymów endonukleazy UV w komórkach pacjenta (w fibroblastach) lub ich całkowity brak odgrywają ważną rolę w rozwoju choroby. Enzym ten odpowiada za reprodukcję DNA uszkodzonego przez promienie ultrafioletowe. Według innych źródeł, niedobór enzymu polimerazy DNA-1 odgrywa ważną rolę w rozwoju choroby u niektórych pacjentów. Choroba rozwija się najczęściej pod wpływem promieni o długości fali 280-310 nm. Uważa się, że kserodermia pigmentowa rozwija się z powodu wnikania do organizmu różnych substancji fotodynamicznych lub wzrostu porfiryn w środowisku biologicznym człowieka.
We wczesnych stadiach choroby obserwuje się hiperkeratozę, zanik ognisk naskórka, ścieńczenie warstwy Malpighiego, wzrost granulek melaniny w warstwie podstawnej komórek oraz przewlekłe nacieki zapalne w górnej warstwie skóry właściwej (głównie wokół naczyń krwionośnych). Następnie wykrywa się zmiany zwyrodnieniowe włókien kolagenowych i elastycznych, a w stadium guza ujawniają się objawy charakterystyczne dla raka skóry.
Objawy xeroderma pigmentosum
Mężczyźni i kobiety są w równym stopniu dotknięci tą chorobą. Choroba rozpoczyna się we wczesnym dzieciństwie wiosną lub latem. Skóra pacjenta jest szczególnie wrażliwa na światło słoneczne. Dlatego pierwsza wysypka w postaci hiperpigmentacji, podobnej do pieprzyka, pojawia się na obszarach skóry wystawionych na działanie promieni słonecznych. Wysypka nasila się, rumień nasila się, staje się ciemnobrązowy, pojawiają się teleangiektazje, naczyniaki i zaniki. Następnie rosną brodawki i kurzajki (głównie na skórze twarzy i szyi), pojawiają się plamy i owrzodzenia. W wyniku bliznowacenia owrzodzenia nos staje się cieńszy („ptasi dziób”), powieka się wywija. Brodawki zwykle rozwijają się w nowotwory złośliwe. Krótka skóra barwnikowa z reguły występuje razem z rakiem kolczystokomórkowym, mięsakami czerniaka.
Co Cię dręczy?
Gradacja
W przebiegu klinicznym skóry pergaminowej wyróżnia się pięć stadiów: stan zapalny (rumieniowy), hiperpigmentację, zanik, nadmierne rogowacenie i nowotwór złośliwy.
W fazie zapalnej (rumieniowej) obrzęk, czerwone plamy, a czasami pęcherze i pęcherzyki pojawiają się na obszarach skóry wystawionych na działanie promieni słonecznych (twarz, szyja, górna część klatki piersiowej, ramiona, dłonie). Na obszarach nie wystawionych na działanie promieni słonecznych nie występuje prawie żadna wysypka.
W przypadku hiperpigmentacji, w miejscu czerwonych plam pojawiają się jasnobrązowe, brązowe, hiperpigmentowane plamy podobne do znamion.
Przy zaniku skóra wysycha, staje się cieńsza, pojawiają się zmarszczki. Na skórze warg i nosa obserwuje się liczne, drobne, gwiaździste teleangiektazje i blizny o błyszczącej powierzchni. Z powodu zaniku i blizn rozwija się mikrotomia (zmniejszenie otworu ustnego), ektropium, ścieńczenie uszu i nosa, atrezja otworu nosowego i ust. U 80-85% pacjentów dotknięte są oczy: obserwuje się zapalenie spojówek, zapalenie rogówki, uszkodzenie rogówki i błony śluzowej oraz osłabienie widzenia. Na skórze powiek obserwuje się dyschromię, teleangiektazje, zmiany hiperkeratotyczne i guzy.
Przy hiperkeratozie w opisanych powyżej ogniskach patologicznych pojawiają się guzy brodawkowate, brodawczaki, keratoacanthoma, włókniaki, naczyniakowłókniaki i inne łagodne nowotwory. Dlatego niektórzy naukowcy zaliczają kserodermę barwnikową do grupy chorób przednowotworowych.
Po 10-15 latach od wystąpienia choroby złośliwe nowotwory skóry (basalioma, endothelioma, angiosarcoma) pojawiają się w ogniskach zanikowych i plamach barwnikowych. Guzy ulegają zniszczeniu w krótkim czasie, dają przerzuty do narządów wewnętrznych i prowadzą do śmierci. W narządach wewnętrznych i tkankach niektórych pacjentów obserwuje się ogólne zmiany dystroficzne (syndactelia drugiego i trzeciego palca, dystrofia zębów, całkowita utrata włosów itp.).
Formularze
Neurologiczna postać choroby xeroderma pigmentosum objawia się dwoma zespołami chorobowymi.
Zespół Reeda charakteryzuje się klinicznymi objawami xeroderma pigmentosum i małogłowia, idiopatii i powolnym wzrostem układu kostnego. W neurologicznej postaci choroby naprawa DNA jest trudna, a terapia rentgenowska prowadzi do zwiększenia głównych ognisk patologicznych.
Zespół De Sinctis-X Cocchione charakteryzuje się następującymi objawami klinicznymi:
- rozwój objawów klinicznych xeroderma pigmentosum na skórze szczególnie wrażliwej na światło;
- wczesne pojawienie się nowotworów złośliwych;
- jednoczesny przebieg porażenia spastycznego, małogłowia i demencji;
- wady wrodzone i karłowatość;
- niedorozwój gonad;
- częste poronienia;
- dziedziczenie recesywne.
Według niektórych dermatologów zespół Santisa-Cacchione'a nie jest samodzielną chorobą, lecz ciężką i pełnoobjawową manifestacją kliniczną skóry pergaminowej.
Formy genetyczne
Typy |
Gen |
Umiejscowienie |
Opis |
Typ A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) grupa A – forma klasyczna |
Typ B, II, XPB |
XPB |
2 kw. 21 |
Grupa XP B |
Typ C, III, XPC |
XPC |
3p25 |
Grupa XP C |
Typ D, IV, XPD |
XPD-ERCC6 |
19q13.2-q13.3, 10q11 |
XP Grupa D lub zespół De Sanctis-Cacchione |
Typ E, V, XPE |
DDB2 |
11p12-p11 |
Grupa XP E |
Typ F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
Grupa XP F |
Typ G, VII, XPG |
RAD2ERCC5 |
13q33 |
Grupa XP G i zespół COFS (zespół mózgowo-oczno-twarzowo-szkieletowy) typu 3 |
Typ V, XPV |
POLH |
6p21.1-p12 |
Wariant Xeroderma Pigmentosa |
Co trzeba zbadać?
Jak zbadać?
Z kim się skontaktować?
Leczenie xeroderma pigmentosum
Leki przeciwmalaryczne (delagyl, plaquenil, resoquin itp.), chroniące DNA przed promieniowaniem ultrafioletowym, hamują proces depolimeryzacji, zmniejszają wrażliwość (fotosensybilizację) skóry na światło słoneczne. Leki te mają właściwości przeciwzapalne i hiposensybilizujące. Zaleca się prowadzenie terapii ogólnej łącznie z terapią witaminową (B1, B2, PP, B6, B12, A, E), lekami przeciwhistaminowymi (tavegil, difenhydramina, suprastin), środkami odczulającymi (tiosiarczan sodu, 10% chlorek wapnia dożylnie 10 ml).
Do leczenia miejscowego stosuje się kremy i maści z filtrem przeciwsłonecznym.
W przypadku postaci guza kserodermii pigmentowej stosuje się metody chirurgiczne, ciekły azot i wiązki laserowe. Aby chronić się przed słońcem, należy nosić luźne ubrania, kapelusze przeciwsłoneczne i rękawiczki.
Prognoza
Większość pacjentów (2/3) umiera przed ukończeniem 15 roku życia. Niektórzy pacjenci mogą dożyć 40–50 lat.
[ 20 ]