Ekspert medyczny artykułu

Nowe publikacje
Zespół Martina-Bella
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
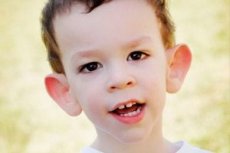
Zespół Martina-Bella został opisany w 1943 roku przez lekarzy, od których pochodzi jego nazwa. Choroba jest chorobą genetyczną polegającą na upośledzeniu umysłowym. W 1969 roku zidentyfikowano zmiany w chromosomie X (kruchość w ramieniu dystalnym) charakterystyczne dla tej choroby. W 1991 roku naukowcy odkryli gen odpowiedzialny za rozwój tej choroby. Choroba ta jest również nazywana „zespołem łamliwego chromosomu X”. Zarówno chłopcy, jak i dziewczynki są podatni na tę chorobę, ale chłopcy są dotknięci częściej (3 razy).
Epidemiologia
Zespół Martina-Bella jest dość powszechną chorobą: 0,3-1,0 na 1000 mężczyzn cierpi na tę chorobę, a 0,2-0,6 na 1000 kobiet. Ponadto dzieci z zespołem Martina-Bella rodzą się na wszystkich kontynentach z taką samą częstością. Oczywistym jest, że narodowość, kolor skóry, kształt oczu, warunki życia i samopoczucie ludzi nie mają wpływu na występowanie choroby. Częstotliwość jej występowania jest porównywalna jedynie z częstością występowania zespołu Downa (1 choroba na 600-800 noworodków). Piąta część męskich nosicieli zmienionego genu jest zdrowa, nie ma żadnych nieprawidłowości klinicznych ani genetycznych, reszta ma objawy upośledzenia umysłowego od postaci łagodnej do ciężkiej. Wśród nosicielek płci żeńskiej nieco ponad jedna trzecia jest chora.
Zespół łamliwego chromosomu X dotyka około 1 na 2500–4000 mężczyzn i 1 na 7000–8000 kobiet. Częstość występowania nosicielstwa wśród kobiet szacuje się na 1 na 130–250; częstość występowania nosicielstwa wśród mężczyzn szacuje się na 1 na 250–800.
Przyczyny Zespół Martina-Bella
Zespół Martina-Bella rozwija się z powodu całkowitego lub częściowego zaprzestania produkcji określonego białka przez organizm. Dzieje się tak z powodu braku odpowiedzi genu FMR1, zlokalizowanego w chromosomie X. Mutacja występuje w wyniku restrukturyzacji genu z niestabilnych wariantów strukturalnych stanów genu (alleli), a nie od samego początku. Choroba jest przenoszona tylko w linii męskiej, a mężczyzna niekoniecznie musi być chory. Nosiciele płci męskiej przekazują gen swoim córkom w niezmienionej formie, więc ich upośledzenie umysłowe nie jest oczywiste. Przy dalszym przekazywaniu genu od matki do jej dzieci gen mutuje i pojawiają się wszystkie objawy charakterystyczne dla tej choroby.
Patogeneza
Patogeneza zespołu Martina-Bella opiera się na mutacjach aparatu genowego, które prowadzą do zablokowania produkcji białka FMR, białka niezbędnego dla organizmu, zwłaszcza w neuronach, i występującego w różnych tkankach. Badania pokazują, że białka FMR są bezpośrednio zaangażowane w procesy regulacji translacji zachodzące w tkance mózgowej. Brak tego białka lub jego ograniczona produkcja przez organizm prowadzi do upośledzenia umysłowego.
W patogenezie choroby hipermetylacja genu uważana jest za kluczowe zaburzenie, ale do tej pory nie udało się jednoznacznie ustalić mechanizmu rozwoju tego zaburzenia.
Jednocześnie odkryto również heterogeniczność locus patologii, która jest związana z poliallelizmem, a także polilocus. Określono obecność wariantów allelicznych rozwoju choroby, które są spowodowane istnieniem mutacji punktowych, a także zniszczeniem genu typu FMRL.
U pacjentów występują również 2 kruche triplety wrażliwe na kwas foliowy, zlokalizowane 300 kb, a także 1,5-2 mln pb od kruchego tripletu zawierającego gen FMR1. Mechanizm mutacji zachodzących w genach FRAXE i FRAXF (są one identyfikowane w wyżej wymienionych kruchych tripletach) jest związany z mechanizmem zaburzeń w zespole Martina Bella. Mechanizm ten jest spowodowany rozprzestrzenianiem się powtórzeń GCC i CGG, które powodują metylację tzw. wysp CpG. Oprócz klasycznej postaci patologii występują również 2 rzadkie typy, które różnią się ze względu na ekspansję powtórzeń trinukleotydowych (w mejozie męskiej i żeńskiej).
Stwierdzono, że w klasycznej postaci zespołu u pacjenta brakuje specjalnego białka nukleocytoplazmatycznego typu FMR1, które pełni funkcję wiązania różnych mRNA. Ponadto białko to sprzyja tworzeniu kompleksu, który pomaga przeprowadzać procesy translacji wewnątrz rybosomów.
Objawy Zespół Martina-Bella
Jak rozpoznać chorobę u dzieci? Jakie są pierwsze objawy? W pierwszych miesiącach życia dziecka nie sposób rozpoznać objawu Martina-Bella, poza tym, że czasami obserwuje się spadek napięcia mięśniowego. Po roku obraz kliniczny choroby jest bardziej oczywisty: dziecko zaczyna chodzić i mówić późno, czasami mowa jest całkowicie nieobecna. Jest nadpobudliwe, macha rękami bez ładu i składu, boi się tłumów i hałasu, jest uparte, zdarzają się gwałtowne wybuchy złości, jest niestabilne emocjonalnie, występują napady padaczkowe, nie nawiązuje kontaktu wzrokowego. U pacjentów z zespołem Martina-Bella chorobę zdradza również ich wygląd: uszy są odstające i duże, czoło jest ciężkie, twarz wydłużona, broda wysunięta, zez, szerokie dłonie i stopy. Charakteryzują się również zaburzeniami endokrynologicznymi: często duża masa ciała, otyłość, duże jądra u mężczyzn, wczesne dojrzewanie.

Wśród pacjentów z zespołem Martina-Bella poziom inteligencji jest bardzo zróżnicowany: od łagodnego upośledzenia umysłowego do ciężkich przypadków. Jeśli osoba normalna ma średnio iloraz inteligencji (IQ) 100, a geniusz 130, to osoby podatne na tę chorobę mają 35-70.
Wszystkie objawy kliniczne tej patologii można scharakteryzować za pomocą triady głównych objawów:
- oligofrenia (IQ 35-50);
- dysmorfofobia (obserwowano odstające uszy i prognatyzm);
- makroorchidyzm, który pojawia się po rozpoczęciu dojrzewania.
U około 80% pacjentów występuje również wypadanie płatków zastawki dwupłatkowej.
Jednakże pełna forma zespołu ujawnia się tylko u 60% wszystkich pacjentów. U 10% wykrywa się jedynie upośledzenie umysłowe, a u pozostałych choroba rozwija się z inną kombinacją objawów.
Do pierwszych objawów choroby, które pojawiają się już we wczesnym wieku, zalicza się:
- u chorego dziecka stwierdza się znaczne upośledzenie umysłowe w porównaniu z rozwojem jego rówieśników;
- zaburzenia uwagi i koncentracji;
- silna upartość;
- dzieci zaczynają chodzić i mówić dość późno;
- obserwuje się nadpobudliwość psychoruchową i zaburzenia rozwoju mowy;
- bardzo silne i niekontrolowane napady gniewu;
- może rozwinąć się mutyzm – całkowity brak mowy u dziecka;
- dziecko odczuwa lęk społeczny i jest zdolne do paniki z powodu głośnego hałasu lub innych głośnych dźwięków;
- dziecko macha rączkami w sposób niekontrolowany i chaotyczny;
- obserwuje się nieśmiałość, dziecko boi się przebywać w zatłoczonych miejscach;
- pojawianie się różnego rodzaju natrętnej myśli, niestabilny stan emocjonalny;
- Dziecko może być niechętne do nawiązywania kontaktu wzrokowego z ludźmi.
U osób dorosłych obserwuje się następujące objawy patologii:
- wygląd specyficzny: wydłużona twarz z ciężkim czołem, dużymi odstającymi uszami, mocno wysuniętą brodą;
- płaskostopie, zapalenie ucha i zez;
- okres dojrzewania następuje dość wcześnie;
- może rozwinąć się otyłość;
- W zespole Martina-Bella dość często obserwuje się wady serca;
- u mężczyzn obserwuje się powiększenie jąder;
- stawy stają się bardzo ruchome;
- waga i wzrost gwałtownie wzrastają.
Diagnostyka Zespół Martina-Bella
Aby zdiagnozować zespół Martina Bella, należy skontaktować się z wykwalifikowanym genetykiem. Diagnozę stawia się po przeprowadzeniu określonych testów genetycznych, które pozwalają zidentyfikować wadliwy chromosom.
Testy
Na wczesnym etapie choroby stosuje się metodę cytogenetyczną, w której pobiera się od pacjenta fragment materiału komórkowego, do którego dodaje się kwas foliowy, aby wywołać zmiany w chromosomach. Po pewnym czasie identyfikuje się obszar chromosomu, w którym występuje zauważalne przerzedzenie - jest to oznaka obecności zespołu łamliwego chromosomu X.
Jednakże test ten nie nadaje się do diagnozy w późniejszych stadiach choroby, ponieważ jego dokładność jest ograniczona ze względu na powszechne stosowanie preparatów multiwitaminowych zawierających kwas foliowy.
Zintegrowana diagnostyka zespołu Martina-Bella to badanie genetyki molekularnej, które polega na określeniu liczby tzw. powtórzeń trinukleotydów w genie.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnostyka instrumentalna
Wysoce specyficzną metodą diagnostyki instrumentalnej jest PCR (reakcja łańcuchowa polimerazy), która pozwala na zbadanie struktury reszt aminokwasowych zawartych w chromosomie X i na tej podstawie stwierdzenie obecności zespołu Martina Bella.
Istnieje również odrębna, jeszcze bardziej specyficzna metoda diagnostyki patologii – połączenie PCR i detekcji za pomocą elektroforezy kapilarnej. Ta metoda jest bardzo dokładna i wykrywa patologię chromosomalną u pacjentów z pierwotną niewydolnością jajników, a także zespołem ataksji.
Obecność wady można stwierdzić po przeprowadzeniu diagnostyki na podstawie EEG. Pacjenci z tą chorobą mają podobną aktywność bioelektryczną mózgu.
Jakie testy są potrzebne?
Diagnostyka różnicowa
Do zróżnicowanych metod, które pomagają podejrzewać ten zespół chorobowy, zalicza się:
- kliniczne - u 97,5% chorych występują wyraźne objawy upośledzenia umysłowego (umiarkowanego lub głębokiego); u 62% odstające duże uszy; u 68,4% duży, wystający podbródek i czoło; u 68,4% chłopców powiększone jądra, u 41,4% zaburzenia mowy (nierówne tempo mowy, niekontrolowana głośność itp.);
- cytogenetyczne - wykonuje się badanie krwi pod kątem hodowli limfocytów, określa się liczbę komórek z łamliwym chromosomem X przypadającą na 100 badanych komórek;
- Elektroencefalografia – rejestruje się zmiany w impulsach elektrycznych mózgu charakterystyczne dla zespołu Martina-Bella.
Z kim się skontaktować?
Leczenie Zespół Martina-Bella
W leczeniu pacjentów dorosłych stosuje się leki przeciwdepresyjne z psychostymulantami. Proces farmakoterapii jest stale monitorowany przez psychologa i psychiatrę. Ponadto w prywatnych klinikach wykonuje się zabiegi mikroiniekcji lekami takimi jak Cerebrolysin (lub jego pochodne), a także cytomedyny (takie jak Solcoseryl lub Lidase).
W rozwoju zespołu ataksji stosuje się leki rozrzedzające krew i nootropiki. Ponadto przepisuje się mieszanki aminokwasów i angioprotektory. Kobietom z pierwotną niewydolnością jajników przepisuje się leczenie korygujące z użyciem leków ziołowych i estrogenów.
W leczeniu stosuje się również antagonistów receptora glutaminy.
Tradycyjnie leczenie zespołu Martina-Bella polega na stosowaniu leków, które wpływają na objawy choroby, ale nie na jej przyczynę. Terapia ta polega na przepisywaniu leków przeciwdepresyjnych, neuroleptyków i psychostymulantów. Nie wszystkie leki są wskazane do stosowania u dzieci, więc lista leków jest dość ograniczona. Neuroleptyki, które można stosować po 3 latach (najwcześniejszy wiek na ich przepisanie) obejmują haloperydol w kroplach i tabletkach, chloropromazynę w roztworze i perycjazynę w kroplach. Tak więc dawka haloperidolu dla dzieci jest obliczana w zależności od masy ciała. Dla dorosłych dawka jest przepisywana indywidualnie. Przyjmuje się ją doustnie, zaczynając od 0,5–5 mg 2–3 razy dziennie, następnie dawkę stopniowo zwiększa się do 10–15 mg. Gdy nastąpi poprawa, przechodzą na niższą dawkę, aby utrzymać osiągnięty stan. W przypadku pobudzenia psychoruchowego przepisuje się 5–10 mg domięśniowo lub dożylnie, możliwe jest kilkakrotne powtórzenie po 30–40 minutach. Dawka dzienna nie powinna przekraczać 100 mg. Możliwe są działania niepożądane w postaci nudności, wymiotów, skurczów mięśni, wzrostu ciśnienia, arytmii itp. Osoby starsze powinny zachować szczególną ostrożność, ponieważ odnotowano przypadki nagłego zatrzymania akcji serca i mogą wystąpić późne dyskinezy (mimowolne ruchy).
Leki przeciwdepresyjne zwiększają aktywność struktur mózgowych, łagodzą depresję, napięcie i poprawiają nastrój. Leki te, zalecane do stosowania od 5 do 8 roku życia w zespole Martina-Bella, obejmują klomiprominę, sertralinę, fluoksetynę i fluwoksaminę. Tak więc fluoksetynę przyjmuje się doustnie podczas posiłków 1-2 razy (najlepiej w pierwszej połowie dnia), zaczynając od 20 mg na dobę, zwiększając do 80 mg, jeśli to konieczne. Osobom starszym nie zaleca się dawki większej niż 60 mg. Przebieg leczenia ustala lekarz, ale nie dłużej niż 5 tygodni.
Możliwe działania niepożądane: zawroty głowy, lęk, szumy uszne, utrata apetytu, tachykardia, obrzęki itp. Należy zachować ostrożność przepisując lek osobom w podeszłym wieku, cierpiącym na choroby układu krążenia i cukrzycę.
Psychostimulanty to leki psychotropowe stosowane w celu poprawy percepcji bodźców zewnętrznych: wyostrzają słuch, reakcje i wzrok.
Diazepam jest przepisywany jako środek uspokajający na nerwice, lęki, napady padaczkowe i drgawki. Przyjmuje się go doustnie, dożylnie, domięśniowo, doodbytniczo (do odbytu). Jest przepisywany indywidualnie, w zależności od ciężkości choroby, najmniejszymi dawkami 5-10 mg, dziennie - 5-20 mg. Czas trwania leczenia wynosi 2-3 miesiące. U dzieci dawkę oblicza się biorąc pod uwagę masę ciała i cechy indywidualne. Działania niepożądane obejmują letarg, apatię, senność, nudności, zaparcia. Niebezpieczne jest łączenie z alkoholem, możliwe jest uzależnienie od leku.
W leczeniu zespołu Martina-Bella odnotowano przypadki poprawy stanu i wprowadzenia leków wykonanych z materiału zwierzęcego (mózgu): cerebrolysat, cerebrolysin, cerebrolysate-M. Głównymi składnikami tych leków są peptydy, które promują produkcję białka w neuronach, uzupełniając w ten sposób brakujące białko. Cerebrolysin podaje się w postaci strumienia 5-10 ml, cykl leczenia składa się z 20-30 zastrzyków. Lek przepisuje się dzieciom od pierwszego roku życia, podaje się domięśniowo codziennie 1-2 ml przez miesiąc. Możliwe są powtarzane sesje podawania. Działania niepożądane w postaci gorączki, przeciwwskazane dla kobiet w ciąży.
Podejmowano próby leczenia choroby kwasem foliowym, ale poprawiał się tylko aspekt behawioralny (zmniejszał się poziom agresji i nadpobudliwości, poprawiała się mowa), a na poziomie intelektualnym nic się nie zmieniło. Aby poprawić stan choroby, zaleca się kwas foliowy, wskazane są metody fizjoterapii, logopedia, korekta pedagogiczna i społeczna.
Preparaty litu są również uważane za skuteczne, ponieważ pomagają poprawić adaptację pacjenta do środowiska społecznego, a także aktywność poznawczą. Ponadto regulują również jego zachowanie w społeczeństwie.
Stosowanie ziół w zespole Martina-Bella jest możliwe jako środki przeciwdepresyjne. Zioła, które pomagają złagodzić napięcie, lęk i poprawić sen, to kozłek lekarski, mięta pieprzowa, tymianek, dziurawiec i rumianek. Napary przygotowuje się w następujący sposób: na 1 łyżeczkę suchych ziół potrzebna będzie szklanka wrzącej wody, wywary zaparza się przez co najmniej 20 minut, przyjmuje się głównie wieczorem przed snem lub po południu. Dobrym dodatkiem do nich będzie łyżeczka miodu.
Leczenie fizjoterapeutyczne
Aby wyeliminować objawy neurologiczne, stosuje się specjalne zabiegi fizjoterapeutyczne – takie jak ćwiczenia w basenie, relaksacja mięśni i akupunktura.
Leczenie chirurgiczne
Ważnym etapem leczenia są również metody chirurgii plastycznej - operacje, które pomagają poprawić wygląd pacjenta. Wykonuje się operacje plastyczne kończyn i małżowin usznych, a także narządów płciowych. Wykonuje się również korekcję ginekomastii z epispadias, a także innych defektów wyglądu.
Zapobieganie
Jedyną metodą zapobiegania chorobie jest prenatalne badanie przesiewowe kobiet w ciąży. Istnieją specjalne badania, które pozwalają na wczesne wykrycie patologii, po czym zaleca się przerwanie ciąży. Alternatywnie stosuje się zapłodnienie in vitro, które może pomóc dziecku odziedziczyć zdrowy chromosom X.
Profilaktyka u pacjenta zależy od tego, czy mutacja genu pojawiła się ponownie, czy została odziedziczona. W tym celu przeprowadza się diagnostykę genetyczną molekularną. Fakt, że badanie nie wykazało „kruchego chromosomu X” u krewnych, przemawia za „świeżością” mutacji, co oznacza, że ryzyko urodzenia dziecka z zespołem Martina-Bella jest bardzo małe. W rodzinach, w których są osoby chore, badanie pomoże uniknąć nawrotów.
Prognoza
Rokowanie w zespole Martina-Bella jest korzystne dla życia, ale nie dla powrotu do zdrowia. Długość życia zależy od ciężkości choroby i towarzyszących jej defektów. Pacjent może żyć normalnie. W ciężkich postaciach zespołu Martina-Bella pacjenci są narażeni na dożywotnią niepełnosprawność.
Długość życia
Zespół Martina Bella nie ma poważnego negatywnego wpływu na zdrowie, dlatego średnia długość życia większości osób, u których zdiagnozowano tę patologię, nie odbiega od standardowych wskaźników.