
Zespół Martina-Bella
Ostatnia recenzja: 23.04.2024

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
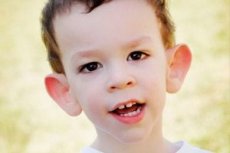
Zespół Martin-Bella został opisany w 1943 roku. Lekarzy, których imiona zostały nazwane. Choroba jest zaburzeniem genetycznym, polegającym na upośledzeniu umysłowym. W 1969 roku zmiany charakterystyczne tej choroby w chromosomie X (kruchość w dystalnym ramieniu) zostały ujawnione. W 1991 roku Naukowcy odkryli gen odpowiedzialny za rozwój tej choroby. Ta dolegliwość jest również nazywana "zespołem kruchego chromosomu X". Choroba dotyka zarówno chłopców, jak i dziewczęta, ale częściej (3 razy) chłopcy chorują.
 [
[Epidemiologia
Zespół Martina-Bella to dość powszechna choroba: na tę przypadłość cierpi 0,3-1,0 osób na 1000 mężczyzn i 0,2-0,6 na 1000 kobiet. A dzieci z zespołem Martina-Bella rodzą się na wszystkich kontynentach z tą samą częstotliwością. Oczywiście narodowość, kolor skóry, nacięcie oczu, warunki życia, dobrostan ludzi nie wpływają na początek choroby. Częstotliwość występowania jest porównywalna tylko z częstością zespołu Downa (dla 600-800 noworodków, 1 choroba). Jedna piąta męskich nosicieli zmienionego genu jest zdrowa, nie ma żadnych klinicznych i genetycznych nieprawidłowości, a reszta ma oznaki upośledzenia umysłowego od łagodnych do ciężkich postaci. Wśród żeńskich nosicieli pacjentów nieco ponad jedna trzecia.
Zespół łamliwego chromosomu X dotyka około 1 na 2500-4000 mężczyzn i 1 na 7000-8000 kobiet. Częstość występowania nosicieli choroby wśród populacji kobiet szacuje się na 1 na 130-250 osób; Częstość występowania nosicieli wśród mężczyzn szacuje się na 1 na 250-800.
Przyczyny zespół Martina-Bella
Zespół Martin-Bella rozwija się z powodu całkowitego lub częściowego zaprzestania produkcji określonego białka przez organizm. Wynika to z braku odpowiedzi z genu, takiego jak FMR1, zlokalizowanego w chromosomie X. Mutacja występuje w wyniku przegrupowania struktury genu z niestabilnych strukturalnych wariantów stanów genowych (alleli), a nie od samego początku. Choroba jest przenoszona tylko przez linię męską, z którą mężczyzna niekoniecznie musi być chory. Męscy nosiciele przekazują gen córkom w niezmienionej postaci, więc ich upośledzenie umysłowe nie jest oczywiste. Wraz z dalszym transferem genu od matki do jej dzieci, gen ulega mutacjom, a następnie pojawiają się wszystkie objawy charakterystyczne dla tej choroby.
Patogeneza
W sercu patogenezy zespołu Martina-Bella znajdują się mutacje w aparacie genowym, które prowadzą do blokowania produkcji białka FMR, białka istotnego dla organizmu, szczególnie w neuronach, i jest obecne w różnych tkankach. Badania wykazują, że białka FMR są bezpośrednio zaangażowane w regulację translacji zachodzących w tkankach mózgu. Brak tego białka lub jego ograniczona produkcja przez organizm prowadzi do upośledzenia umysłowego.
W patogenezie choroby kluczowym naruszeniem jest hipermetylacja genu, ale nie udało się ostatecznie określić mechanizmu rozwoju tego zaburzenia.
Wraz z tym ujawniono również heterogeniczność miejsca patologii, która wiąże się z poliallizmem, jak również polilocieniem. Stwierdzono obecność allelicznych wariantów rozwoju choroby, które są spowodowane przez obecność mutacji punktowych, a także zniszczenie genu FMRL.
Również u pacjentów wykryto 2 wrażliwe na tryplety wrażliwe na kwas foliowy znajdujące się w Z00 kb, a także 2, 2-2 mp. Z kruchego trójki, która zawiera gen FMR1. Mechanizm genów FRAXE, jak również FRAXF (są one zidentyfikowane w wyżej wspomnianych potrójnych tripletach) mutacje korelują z mechanizmem zaburzeń w zespole Martina Bella. Mechanizm ten wynika z propagacji GCC-, a także powtórzeń CGG, w których zachodzi metylacja tak zwanych wysp CpG. Oprócz klasycznej formy patologii istnieją również 2 rzadkie typy, które różnią się ze względu na ekspansję powtórzeń trinukleotydowych (w męskiej i żeńskiej mejozie).
Stwierdzono, że w klasycznej postaci zespołu pacjent nie ma specjalnego białka nukleocytoplazmatycznego typu FMR1, który pełni funkcję wiązania różnych mRNA. Ponadto białko to promuje tworzenie kompleksu, który pomaga w przeprowadzaniu procesów translacyjnych wewnątrz rybosomu.
Objawy zespół Martina-Bella
Jak rozpoznać chorobę u dzieci? Jakie są pierwsze znaki? W pierwszych miesiącach życia dziecka objawy Martina-Bella nie są rozpoznawane, z wyjątkiem tego, że czasami dochodzi do zmniejszenia napięcia mięśniowego. Po roku klinika choroby jest bardziej oczywista: dziecko zaczyna chodzić późno i rozmawiać, czasem mowa jest całkowicie nieobecna. Jest nadpobudliwy, losowo macha rękami, boi się tłumów i hałasu, uparty, pojawiają się gwałtowne wybuchy gniewu, niestabilność emocjonalna, pojawia się napad padaczkowy, nie ma kontaktu wzrokowego. U pacjentów z zespołem Martina-Belli choroba wywołuje również wrażenie: uszy wystające i duże, czoło ciężkie, twarz wydłużona, wystający podbródek, strabismus, szerokie szczotki i stopy. Charakterystyczne są także zaburzenia endokrynologiczne: często ciężka waga, otyłość, mężczyźni mają duże jądra, wczesne dojrzewanie płciowe.

Wśród pacjentów z zespołem Martina-Bella poziom inteligencji jest bardzo różny: od niewielkiego upośledzenia umysłowego do ciężkich przypadków. Jeśli iloraz inteligencji normalnej osoby wynosi średnio 100, a geniusz 130, to osoby dotknięte chorobą mają 35-70.
Wszystkie kliniczne objawy patologii można scharakteryzować za pomocą triady podstawowych objawów:
- oligofrenia (IQ wynosi 35-50);
- dysmorfofobia (wystające uszy, a także prognatyzm);
- makroorchidyzm, który objawia się po rozpoczęciu dojrzewania.
Około 80% pacjentów ujawnia również wypadnięcie zastawki dwudzielnej.
Ale pełna postać tego syndromu objawia się tylko u 60% wszystkich pacjentów. W 10% stwierdza się jedynie upośledzenie umysłowe, podczas gdy w innych choroba rozwija się z inną kombinacją objawów.
Wśród pierwszych oznak choroby objawia się już w młodym wieku:
- dziecko chore ma znaczne upośledzenie umysłowe w porównaniu z rozwojem innych rówieśników;
- zaburzenia uwagi i skupienia;
- silny upór;
- dzieci dość późno zaczynają chodzić i rozmawiać;
- występują nadpobudliwość i zaburzenia w rozwoju mowy;
- bardzo silne i niekontrolowane ataki gniewu;
- może rozwinąć mutizm - jest to całkowity brak mowy dziecka;
- dziecko odczuwa niepokój społeczny, jest w stanie panikować z powodu głośnego hałasu lub innych silnych dźwięków;
- dziecko niekontrolowanie i chaotycznie macha rękami;
- jest nieśmiałość, dziecko boi się przebywać w miejscach dużego tłumu ludzi;
- pojawienie się różnych obsesji, niestabilny stan emocjonalny;
- dziecko może niechętnie nawiązywać kontakt wzrokowy z ludźmi.
U dorosłych obserwuje się następujące objawy patologii:
- specyficzny wygląd: wydłużona twarz z ciężkim czołem, duże wystające uszy, mocno wystający podbródek;
- płaskie stopy, zapalenie ucha i strabismus;
- dojrzewanie występuje dość wcześnie;
- otyłość może się rozwijać;
- dość często w zespole Martina Bella występują wady rozwojowe serca;
- u mężczyzn występuje wzrost liczby jąder;
- stawy stawów stają się bardzo mobilne;
- Waga, a także gwałtownie rośnie.
Diagnostyka zespół Martina-Bella
Aby zdiagnozować zespół Martina Bella, należy skontaktować się z wykwalifikowanym genetykiem. Diagnozę przeprowadza się po wykonaniu specyficznych testów genetycznych, które pozwalają określić wadliwy chromosom.
Analizy
We wczesnym stadium rozwoju choroby stosuje się metodę cytogenetyczną, w której pacjent pobiera kawałek materiału komórkowego, do którego następnie dodaje się kwas foliowy w celu wywołania zmian w chromosomach. Po pewnym czasie ujawnia się region chromosomu, na którym obserwuje się zauważalne przerzedzanie - jest to oznaką obecności kruchego zespołu chromosomowego X.
Ale ta analiza nie jest odpowiednia do diagnozy w późnych stadiach choroby, ponieważ jej dokładność jest mniejsza ze względu na szerokie zastosowanie multiwitamin zawierających kwas foliowy.
Zintegrowana diagnoza zespołu Martina-Bella to badanie genetyczne molekularne polegające na określeniu liczby tak zwanych powtórzeń trójnukleotydowych w genie.
Diagnostyka instrumentalna
Bardzo specyficzną metodą diagnostyki instrumentalnej jest PCR (reakcja łańcuchowa polimerazy), która pozwala badać strukturę reszt aminokwasowych zawartych w chromosomie X i tym samym określać obecność zespołu Martina Bella.
Istnieje również osobna, jeszcze bardziej szczegółowa metoda diagnozowania patologii - połączenie PCR i wykrywania za pomocą elektroforezy kapilarnej. Metoda ta jest bardzo dokładna i ujawnia patologię chromosomową u pacjentów z pierwotną postacią niewydolności jajników, a także zespołu ataksji.
Określić obecność defektu może być po diagnozie na EEG. U pacjentów z tą chorobą obserwuje się podobną bioelektryczną aktywność mózgu.
Jakie testy są potrzebne?
Diagnostyka różnicowa
Zróżnicowane metody, które pomagają podejrzewać zespół obejmują:
- kliniczne - u 97,5% pacjentów występują wyraźne oznaki upośledzenia umysłowego (umiarkowane lub głębokie); 62% miało wystające duże uszy; 68,4% miało wydatne wydatne czoło i czoło; u 68,4% chłopców - jądra są powiększone, w 41,4% - cechy mowy (tempo mowy jest nierównomierne, głośność jest niekontrolowana itd.);
- cytogenetyczne - krew jest badana pod kątem hodowli limfocytów, określa się liczbę komórek z rozbiciem chromosomu X na 100 badanych komórek;
- elektroencefalografia - zmiany w impulsach elektrycznych mózgu są specyficzne dla zespołu Martina-Bella.
Z kim się skontaktować?
Leczenie zespół Martina-Bella
Leki przeciwdepresyjne z psychostymulantami stosuje się w leczeniu dorosłych pacjentów. Proces terapii lekowej jest stale monitorowany przez psychologa i psychiatrę. Oprócz prywatnych klinikach uruchamiania procedur mikroiniekcji takie leki jak Cerebrolysin (lub jego pochodne), a także cytomedines (takie jak Solkoseril lub Lidaza).
Wraz z rozwojem zespołu ataksji stosuje się leki, które rozrzedzają krew, a także nootropy. Ponadto zaleca się stosowanie mieszanin aminokwasów i angioprotektorów. Kobietom z pierwotną postacią niewydolności jajników przepisuje się leczenie korygujące za pomocą fito leków i estrogenów.
Ponadto w leczeniu stosuje się antagonistów receptorów glutaminy.
Tradycyjnie w leczeniu zespołu Martina-Bella stosuje się leki wpływające na objawy choroby, ale nie na jej przyczynę. Terapia polega na mianowaniu leków przeciwdepresyjnych, neuroleptyków, psychostymulantów. Nie wszystkie leki są wskazane do stosowania u dzieci, dlatego lista leków jest raczej ograniczona. Neuroleptyki, które można stosować po 3 latach (najwcześniejszy wiek ich wizyty) obejmują haloperidol w kroplach i tabletkach, chloropromazynę w roztworze, kroplę pericyazyny. Tak więc dawka przyjmowania haloperydolu dla dzieci jest obliczana w zależności od masy ciała. W przypadku dorosłych dawkę podaje się indywidualnie. Zaakceptowany w środku, zaczynać od 0,5-5 mg 2-3 razy dziennie, następnie dawkę stopniowo zwiększa się do 10-15 mg. Kiedy nastąpi poprawa, przejdź do niższej dawki, aby utrzymać osiągnięty stan. Przy stymulacji psychomotorycznej należy podać domięśniowo lub dożylnie 5-10 mg, możliwe są wielokrotne powtórzenia w ciągu 30-40 minut. Dzienna dawka nie powinna przekraczać 100 mg. Możliwe działania niepożądane w postaci nudności, wymiotów, skurczów, zwiększonego ciśnienia, arytmii itp. Szczególne środki ostrożności powinny być przestrzegane przez osoby starsze. Zgłaszano przypadki nagłego zatrzymania krążenia i może pojawić się późne dyskenezje (wystąpienie mimowolnych ruchów).
Leki przeciwdepresyjne zwiększają aktywność struktur mózgowych, łagodzą depresyjny nastrój, napięcie, podnoszą nastrój. Leki te, zalecane do przyjęcia od 5-8 lat z zespołem Martina-Bella, obejmują klomiprominę, sertralinę, fluokseginę, fluwoksaminę. Zatem fluoksetyna jest przyjmowana podczas posiłków w ciągu 1-2 (najlepiej rano), zaczynając od 20 mg na dzień, w razie potrzeby zwiększając do 80 mg. Starsi ludzie nie zalecają dawki wyższej niż 60 mg. Przebieg leczenia określa lekarz, ale nie więcej niż 5 tygodni.
Możliwe działania niepożądane: zawroty głowy, niepokój, szum w uszach, zmniejszenie apetytu, tachykardia, obrzęk itp. Podczas powoływania osób starszych, z chorobami sercowo-naczyniowymi, cukrzycą należy zachować ostrożność.
Środki psychostymulujące - leki psychotropowe, stosowane w celu zwiększenia percepcji zewnętrznych bodźców: zaostrzają słuch, odpowiedź, widzenie.
Jako środek uspokajający z neurozami zaleca się lęk, napady padaczkowe, drgawki, diazepam. Jest przyjmowany doustnie, dożylnie, domięśniowo, doodbytniczo (do odbytnicy). Jest przepisywany indywidualnie, w zależności od ciężkości choroby, od najmniejszych dawek 5-10 mg, codziennie - 5-20 mg. Czas leczenia wynosi 2-3 miesiące. W przypadku dzieci dawka jest obliczana z uwzględnieniem masy ciała i indywidualnych cech. Działania niepożądane to letarg, apatia, senność, nudności, zaparcia. Niebezpiecznie jest łączyć się z alkoholem, być może uzależniającym od narkotyku.
Podczas leczenia przypadków Martin Bell-Syndrome poprawy stałe i wprowadzanie produktów wytwarzanych na bazie surowców pochodzenia zwierzęcego (linka): tserebrolizata, Cerebrolysin, tserebrolizat-M. Głównymi składnikami tych leków są peptydy, które przyczyniają się do produkcji białka w neuronach, uzupełniając w ten sposób brakujące białko. Cerebrolizynę wstrzykiwano w ilości 5-10 ml, przebieg leczenia składa się z 20-30 wstrzyknięć. Dzieci przepisują lek od wieku życia, wstrzykuje się domięśniowo każdego dnia za 1-2 ml w ciągu miesiąca. Możliwe powtarzane sesje przyjęć. Skutki uboczne w postaci ciepła, przeciwwskazane u kobiet w ciąży.
Próbowano leczyć dolegliwość kwasem foliowym, ale poprawiono tylko aspekt behawioralny (poziom agresji, obniżono nadpobudliwość, poprawiono mowę) i na poziomie intelektualnym nic się nie zmieniło. Aby poprawić stan choroby zalecany kwas foliowy, wskazane są metody fizjoterapii, logopedyczne, pedagogiczne i społeczne.
Preparaty litu są również uważane za skuteczne, co pomaga poprawić adaptację pacjenta w środowisku społecznym, a także aktywność poznawczą. Ponadto nadal regulują jego zachowanie w społeczeństwie.
Stosowanie ziół w zespole Martina-Bella jest możliwe jako leki przeciwdepresyjne. Zioła, które pomagają złagodzić napięcie, niepokój, poprawiają sen obejmują waleriany, miętę, tymianek, ziele dziurawca, rumianek. Napary przygotowuje się w następujący sposób: 1 łyżeczka suszonych ziół będzie potrzebowała szklanki wrzącej wody, napary nalegają co najmniej 20 minut, pobrane głównie w nocy przed pójściem spać lub po południu. Dobrym dodatkiem do nich będzie łyżka miodu.
Leczenie fizjoterapeutyczne
Aby wyeliminować objawy neurologiczne, wykonywane są specjalne zabiegi fizjoterapii - takie jak ćwiczenia na basenie, rozluźnienie mięśni i akupunktura.
Leczenie operacyjne
Ważnym etapem leczenia są również metody chirurgii plastycznej - operacje, które pomagają poprawić wygląd pacjenta. Plastyk kończyn i małżowin usznych, a poza tym genitalia są wykonywane. Istnieje również korekcja ginekomastii z epispatią, a wraz z nią inne wady wyglądu.
Zapobieganie
Jedyną metodą zapobiegania tej chorobie jest badanie prenatalne kobiet w ciąży. Istnieją specjalne testy, które pozwalają na wczesne wykrycie obecności patologii, po czym zaleca się przerwanie ciąży. Jako alternatywę stosuje się IVF, który może pomóc dziecku w odziedziczeniu zdrowego chromosomu X.
Profilaktyka pacjenta zależy od tego, czy mutacja genu pojawiła się ponownie, czy została odziedziczona. W tym celu przeprowadza się diagnostykę genetyczną molekularną. Na korzyść "świeżości" mutacji fakt, że krewni nie ujawnili "kruchego chromosomu X" świadczy o tym, że ryzyko posiadania dziecka z zespołem Martina-Bella jest bardzo małe. W rodzinach, w których są pacjenci, test pomoże uniknąć powtarzających się przypadków.
Prognoza
Rokowanie zespołu Martina-Bella na całe życie jest korzystne, dla powrotu do zdrowia - nie. Oczekiwana długość życia zależy od ciężkości choroby i towarzyszących mu wad. Pacjent może przeżyć normalne życie. W ciężkich postaciach zespołu Martina-Bella zagrażająca życiu niepełnosprawność zagraża pacjentom.
Długość życia
Zespół Martina Bella nie ma poważnego negatywnego wpływu na zdrowie, więc średnia długość życia większości osób, które znalazły tę patologię, nie różni się od standardowych wskaźników.