
Pigmentowana xeroderma
Ostatnia recenzja: 23.04.2024

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
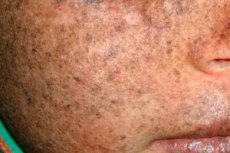
Xeroderma pigmentowa jest autosomalnym recesywnym genetycznym zaburzeniem naprawy DNA. Choroba opiera się na mutacjach w genach, które biorą udział w naprawie uszkodzonego DNA pod wpływem promieniowania ultrafioletowego i innych substancji toksycznych.
Najczęściej choroba ta dotyka dzieci, które są również nazywane "dziećmi nocy". Częstymi powikłaniami choroby są rak podstawnokomórkowy i inne złośliwe nowotwory skóry, przerzutowy złośliwy czerniak i rak płaskonabłonkowy.
 [1],
[1],
Przyczyny pigmentowa xeroderma
Xeroderma pigmentowa jest chorobą dziedziczną przenoszoną przez geny autosomalne, odziedziczone po rodzicach, krewnych i mające charakter rodzinny. Obiad (1967 r.) W siedmioosobowej rodzinie składającej się z siedmiu braci i sióstr ujawnił obecność objawów klinicznych kserkodermy pigmentowej.
Patogeneza
W pojawieniu się choroby ważną rolę odgrywa niedobór enzymów endonukleazy UV w komórkach pacjenta (w fibroblastach) lub jego całkowity brak. Enzym ten jest odpowiedzialny za reprodukcję DNA, na którą wpływa promieniowanie ultrafioletowe. Według innych informacji, u niektórych pacjentów brak enzymu DNA-polimerazy-1 odgrywa ważną rolę w wystąpieniu choroby. Choroba najczęściej rozwija się pod wpływem promieni o długości fali 280-310 nm. Wyrażana jest opinia, że kseroperma pigmentowa rozwija się w wyniku spożycia różnych substancji fotodynamicznych lub porfiryn w ludzkim środowisku biologicznym.
W początkowym okresie choroby obserwowano nadmierne rogowacenie ogniskowej atrofii naskórka, ścieńczenie malpighian warstwy zwiększona granulki melaniny w warstwie podstawowej komórki, przewlekłego nacieku zapalnego w górnej skórze właściwej (głównie wokół naczyń krwionośnych). Później włókna kolagenowe i elastyczne ujawniają zmiany zwyrodnieniowe, a na etapie nowotworów - objawy charakterystyczne dla raka skóry.
Objawy pigmentowa xeroderma
Mężczyźni i kobiety są dotknięci tą chorobą w ten sam sposób. Choroba zaczyna się najwcześniej w dzieciństwie wiosną lub latem. W tym samym czasie skóra pacjenta jest szczególnie wrażliwa na światło słoneczne. Dlatego na obszarach skóry dotkniętych światłem słonecznym pojawia się pierwsza wysypka w postaci przebarwienia podobna do kretu. Wysypka wzrasta, rumień nasila się, staje się ciemnobrązowy, pojawia się teleangiektazja, naczyniak i atrofia. W późniejszym rozwoju brodawczaka i brodawek (głównie na skórze twarzy, szyi) pojawiają się plamy i owrzodzenia. W wyniku powstawania blizn owrzodzenia, nos staje się cieńszy ("dziób ptaka"), pojawia się powieka. Zwykle brodawczaki zamieniają się w nowotwory złośliwe. Xeroderma pigmentowa występuje z reguły razem z rakiem kręgosłupa, melanosarcomą.
Co Cię dręczy?
Gradacja
W przebiegu klinicznym kseroterapii pigmentowej wyróżnia się pięć etapów: zapalny (rumieniowaty), przebarwienia, atrofia, nadmierne rogowacenie i nowotwory złośliwe.
W stadium zapalnym (rumieniowym) obrzęki, czerwone plamki, a czasem bąbelki i pęcherzyki pojawiają się na obszarach skóry narażonych na działanie światła słonecznego (twarz, szyja, górna klatka piersiowa, dłonie, dłonie). W obszarach, które nie są wystawione na działanie promieni słonecznych, prawie nie ma wysypki.
Kiedy przebarwienia w miejscu czerwonych plam pojawiają się podobne do znamion jasnobrązowych, brązowych przebarwień.
Z atrofią skóra wysycha, rozrzedza się, zmarszczki. Na skórze warg i nosa znajduje się wiele małych, gwiaździstych teleangiektazji, blizn o błyszczącej powierzchni. Z powodu atrofii i blizn rozwija się mikrotomia (zmniejszenie otworu w jamie ustnej), ektopia, przerzedzenie uszu i nosa, atrezja otworu nosa i ust. U 80-85% pacjentów dotyczy to oczu: zapalenie spojówek, zapalenie rogówki, uszkodzenie rogów i błony śluzowej, osłabienie widzenia. Dyschromia, teleangiektazje, zmiany hiperkeratotyczne i guzy obserwuje się na skórze powiek.
W hiperkeratozie, w powyższych patologicznych ogniskach pojawiają się guz brodawki, brodawczak, rogowiak kolczystokomórkowy, włókniak, angiofibrioma i inne łagodne nowotwory. Dlatego niektórzy naukowcy obejmują barwnikowe xerodermy w grupie chorób przedrakowych.
Po 10-15 latach od wystąpienia choroby złośliwe guzy skóry (komórka podstawna, śródbłonek, naczyniakomięsak) pojawiają się w zmienionych atroficznie ogniskach i plamach pigmentowych. Nowotwory w krótkim czasie ulegają zniszczeniu, powodują przerzuty do narządów wewnętrznych i prowadzą do śmierci. W narządach wewnętrznych i tkankach niektórych pacjentów obserwuje się ogólne zmiany dystroficzne (zespół II, trzeci palec, dystrofia zębów, całkowite wypadanie włosów, itp.).
Formularze
Postać neurologiczna pigmentowej kserostermy manifestuje się dwoma zespołami.
W zespole obserwuje się objawy kliniczne kserokmentu barwnikowego i małogłowia, idiopatię, spowolnienie wzrostu szkieletu. W przypadku neurologicznej postaci choroby naprawa DNA jest trudna, a terapia promieniami X prowadzi do zwiększenia głównych patologicznych ognisk.
Zespół Sanctis-Kakkione (De Sinctis-X Cocchione) objawia się następującymi objawami klinicznymi:
- rozwój objawów klinicznych kserotypów pigmentowych na szczególnie światłoczułej skórze;
- wczesne pojawienie się nowotworów złośliwych;
- jednoczesny przebieg spastycznego porażenia, małogłowie i otępienie;
- wrodzone deformacje i karłowatość;
- niedorozwój gonad;
- częste poronienia;
- recesywny transfer przez dziedziczenie.
Według niektórych dermatologów, zespół Santis-Kakkione nie jest chorobą niezależną, ale ciężką i całkowicie wyraźną kliniczną manifestacją pigmentowej xerodermy.
Formy genetyczne
|
Rodzaje |
Gene |
Locus |
Opis |
|
PORADA A, I, XPA |
XPA |
9q22.3 |
Xeroderm pigmentowy (XP) grupy A - forma klasyczna |
|
Tип B, II, XPB |
XPB |
2q21 |
Grupa XP B |
|
Tип C, III, XPC |
XPC |
3p25 |
Grupa XP C |
|
Tип D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP grupy D lub Syndrome De Sanctis - Kakkione |
|
Tis E, V, XPE |
DDB2 |
11p12-p11 |
Grupa XP E |
|
Typ F, VI, XPF |
ERCC4 |
16 p133,3-p13.13 |
Grupa XP F |
|
Typ G, VII, XPG |
RAD2 ERCC5 |
13q33 |
Zespół XP grupy G i zespół COFS (zespół mózgowo-twarzowo-szkieletowy) typu 3 |
|
Wpisz V, XPV |
POLH |
6p21.1-p12 |
Wariant pigmentowy Xeroderma |
Co trzeba zbadać?
Jak zbadać?
Z kim się skontaktować?
Leczenie pigmentowa xeroderma
Leki przeciwmalaryczne (delagil, Plaquenil, rezohin et al.), DNA zabezpieczające przed działaniem promieni ultrafioletowych, hamują proces depolimeryzacji, upośledzenie czułości (nadwrażliwość) skóry na działanie promieni słonecznych. Leki te mają właściwości przeciwzapalne i przeciw podrażnieniom. Korzystnie prowadzi się terapię całkowitą terapii witaminy (B1, B2, PP, B6, B12, A, E), środki przeciwhistaminowe (Tavegilum, difenhydramina, Suprastinum), środki odczulające (tiosiarczan sodu, żyła - 10% roztwór chlorku wapnia, 10 ml ).
Jako zabieg miejscowy stosuje się fotoprotekcyjne kremy i maści.
W postaci guza pigmentowej kserokosty stosuje się metodę chirurgiczną, ciekły azot i wiązkę laserową. Aby chronić przed światłem słonecznym, musisz nosić luźne ubranie, panamy i rękawiczki.
Prognoza
Większość pacjentów (2/3) umiera przed osiągnięciem 15 roku życia. Niektórzy pacjenci mogą żyć do 40-50 lat.
[20]