Ekspert medyczny artykułu

Nowe publikacje
Neuroblastoma u dzieci: przyczyny, diagnoza, leczenie
Ostatnia recenzja: 04.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
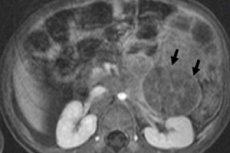
W onkologii dziecięcej jednym z najczęściej występujących nowotworów pozaczaszkowych jest neuroblastoma u dzieci, który jest złośliwym nowotworem zarodkowym neuroblastów grzebienia nerwowego, czyli zarodkowych (niedojrzałych) komórek nerwowych układu współczulnego.
Epidemiologia
Według statystyk Międzynarodowej Grupy Ryzyka Neuroblastoma (INRG), neuroblastoma stanowi około 8% wszystkich chorób onkologicznych u dzieci na całym świecie i jest trzecią najczęstszą chorobą nowotworową, po białaczce i guzach mózgu.
Według innych danych neuroblastoma stanowi około 28% wszystkich nowotworów u niemowląt. Ponad jedna trzecia przypadków neuroblastoma jest diagnozowana u dzieci poniżej pierwszego roku życia; średni wiek diagnozy wynosi 19-22 miesiące. Ponad 90% zdiagnozowanych przypadków występuje u dzieci w wieku od dwóch do pięciu lat (z przewagą chłopców); szczyt zapadalności obserwuje się w wieku od dwóch do trzech lat, a przypadki u dzieci powyżej pięciu lat stanowią mniej niż 10%.
Przyczyny nerwiakowłókniaki
Badając przyczyny neuroblastomy, naukowcy doszli do wniosku, że ten guz u dzieci powstaje z powodu sporadycznych mutacji genetycznych podczas embriogenezy lub wczesnego rozwoju postnatalnego. Jednak nie wiadomo, co powoduje te zmiany genetyczne, ponieważ nie zidentyfikowano wpływu teratogennych czynników środowiskowych.
Tego typu nowotwory mogą pojawić się w dowolnym miejscu, m.in. w śródpiersiu, szyi, jamie brzusznej, nadnerczach, nerkach, kręgosłupie i miednicy.
W rzadkich przypadkach neuroblastoma u niemowląt może być związana z dziedziczną mutacją. W szczególności mutacja w genie białka błonowego CD246 na chromosomie 2 - enzymu kinazy tyrozynowej ALK, który zapewnia komunikację międzykomórkową i odgrywa ważną rolę w funkcjonowaniu układu nerwowego; w genie białka PHOX2B (na chromosomie 4), który bierze udział w dojrzewaniu komórek nerwowych.
Neuroblastoma może być również związana z nerwiakowłókniakowatością wieku dziecięcego typu 1,zespołem Beckwitha-Wiedemanna i hipoglikemią hiperinsulinemiczną (nesidioblastyczne zapalenie trzustki).
Czynniki ryzyka
Obecnie za czynniki ryzyka rozwoju neuroblastoma u dzieci uznaje się dziedziczność - obecność tego guza w historii rodzinnej, a także wrodzone anomalie związane z mutacjami genów w trakcie rozwoju wewnątrzmacicznego. Dotyczy to w szczególności przypadków rozwoju kilku nowotworów w różnych narządach.
Naukowcy nie zidentyfikowali żadnego z czynników zewnętrznych, które zwiększałyby ryzyko wystąpienia tego nowotworu.
Patogeneza
Mechanizm powstawania neuroblastomów jest spowodowany zaburzeniami różnicowania i dojrzewania komórek grzebienia nerwowego – dwustronnych linii komórkowych, które powstają na obrzeżach cewy nerwowej z ektodermalnej warstwy zarodkowej zarodka ludzkiego. Komórki te migrują (poruszają się) i różnicują się w wiele typów komórek: neurony czuciowe i autonomiczne, komórki neuroendokrynne i komórki rdzenia nadnerczy, komórki chrząstki twarzoczaszki i kości, a także komórki barwnikowe.
W przypadku neuroblastomy migrujące neuroblasty nie dojrzewają, ale nadal rosną i dzielą się, tworząc guz. A patogeneza jego powstawania jest związana z następującymi mutacjami genów:
- z duplikacją części sekwencji chromosomowej lub duplikacją segmentów genu LMO1 na chromosomie 11, kodującego białko RBTN1 w komórkach grzebienia nerwowego zarodka;
- ze zmianą liczby kopii genu NBPF10 na chromosomie 1q21.1, kodującego białko DUF1220, które kontroluje proliferację ludzkich komórek macierzystych układu nerwowego. Zaburzenia te prowadzą albo do duplikacji tego chromosomu, albo do jego usunięcia – braku części DNA;
- ze zmianami w genie supresora nowotworu ATRX (na chromosomie Xq21.1);
- z obecnością dodatkowych kopii (amplifikacją) genu czynnika transkrypcyjnego N-Myc na chromosomie 2, który koduje jeden z czynników transkrypcyjnych (białko wiążące DNA), regulujący aktywność innych genów i kontrolujący proliferację komórek prekursorowych podczas tworzenia białek do budowy tkanek i narządów płodu. Amplifikacja tego genu zamienia go w onkogen, co powoduje zaburzenie cyklu komórkowego, zwiększoną proliferację komórek i powstawanie guza.
Objawy nerwiakowłókniaki
Pierwsze objawy neuroblastomy są nieswoiste i mogą obejmować utratę apetytu (i spadek masy ciała), uczucie zmęczenia podczas jedzenia, gorączkę i bóle stawów.
Objawy kliniczne zależą od umiejscowienia guza pierwotnego i obecności przerzutów (które występują w 60-73% przypadków).
Bardzo często pierwotny neuroblastoma jest zlokalizowany w rdzeniu nadnerczy, który ma podobne pochodzenie do komórek nerwowych. U dzieci poniżej pierwszego roku życia neuroblastoma nadnerczy jest diagnozowana w 35-40% przypadków. Jego objawy obejmują ból brzucha, gorączkę, utratę wagi, ból kości, niedokrwistość lub współistniejący zespół Peppera: rozlane uszkodzenie wątroby z ciężką hepatomegalią i zespołem niewydolności oddechowej.
Neuroblastoma zaotrzewnowa, czyli neuroblastoma zaotrzewnowa u dzieci, w miarę wzrostu zaczyna uciskać pęcherz moczowy lub jelita, co może powodować problemy z oddawaniem moczu lub stolca, obrzęki nóg (u chłopców puchnie moszna).
Neuroblastoma śródpiersia u dzieci (neuroblastoma śródpiersia) często uciska żyłę główną górną, co może powodować obrzęk twarzy, szyi, ramion i górnej części klatki piersiowej (skóra staje się niebieskawo-czerwona, z guzkami podskórnymi). Pojawia się kaszel i świszczący oddech, problemy z oddychaniem (duszność) lub problemy z połykaniem (dysfagia); powiększone węzły chłonne są zauważalne na szyi, nad obojczykiem i w pachach.
Rozprzestrzenianie się komórek nowotworowych do szpiku kostnego prowadzi do anemii, trombocytopenii i leukopenii ze skłonnością do krwawień.
A przy przerzutach w okolicy okołooczodołowej wokół oczu pojawiają się cienie lub siniaki. Taki guz może również powodować bóle głowy i zawroty głowy, wytrzeszcz (wytrzeszcz gałek ocznych), a z powodu ucisku zakończeń nerwowych - opadanie powiek (ptoza) i zmniejszenie rozmiaru źrenic (mioza).
Neuroblastoma brzuszna lub neuroblastoma jamy brzusznej u dzieci prowadzi do tworzenia się wyczuwalnych pieczęci w jamie brzusznej, jej wzdęcia, utraty apetytu, zaparć i wzrostu ciśnienia krwi. Guz uciskający rdzeń kręgowy lub korzeń nerwowy może prowadzić do drętwienia i osłabienia kończyn, niemożności stania, raczkowania lub chodzenia. Jeśli kości są dotknięte, może wystąpić ból kości.
W przypadku guza w jamie brzusznej w stadium 3-4 z uszkodzeniem węzłów chłonnych, komórki nowotworowe mogą przedostać się do miąższu nerki, a u dzieci rozwija się wówczas rozległy neuroblastoma nerki, co prowadzi do zaburzenia jej funkcji.
Gradacja
- Neuroblastoma pierwszego stopnia to nowotwór pierwotny, który jest ograniczony i ograniczony do jednej okolicy ciała; węzły chłonne po obu stronach nie są dotknięte.
- Neuroblastoma stadium 2. W stadium 2A guz pierwotny jest ograniczony do jednego obszaru, ale jest duży; nie są zaangażowane węzły chłonne obustronne. W stadium 2B węzły chłonne po stronie ciała, po której znajduje się guz, są dodatnie pod kątem przerzutów.
- Neuroblastoma w stadium 3: guz pierwotny przekracza rdzeń kręgowy lub linię środkową ciała; przerzuty jednostronne lub obustronne występują w węzłach chłonnych.
- Neuroblastoma stadium 4: guz rozprzestrzenił się do odległych węzłów chłonnych, szpiku kostnego, kości, wątroby lub innych narządów. A stadium 4S określa się u dzieci poniżej pierwszego roku życia z zlokalizowanym guzem pierwotnym, z rozsiewem do skóry, wątroby lub szpiku kostnego.
Międzynarodowy System Oceny Ryzyka Neuroblastoma (INRGSS)
INRGSS korzysta z czynników ryzyka zdefiniowanych na podstawie obrazowania (IDRF), czyli czynników widocznych w badaniach obrazowych, które mogą oznaczać, że usunięcie guza będzie trudniejsze.
INRGSS dzieli neuroblastomy na 4 stadia:
- L1: Guz nie rozprzestrzenił się od miejsca, w którym się pojawił, i nie rozrósł się do ważnych struktur. Jest ograniczony do jednej części ciała, takiej jak szyja, klatka piersiowa lub brzuch.
- L2: Guz nie rozprzestrzenił się (nie dał przerzutów) daleko od miejsca początkowego (na przykład mógł rozrosnąć się z lewej strony brzucha na lewą stronę klatki piersiowej), ale ma co najmniej jeden IDRF.
- M: Guz dał przerzuty do odległej części ciała (wyjątkiem są guzy w stadium SM).
- SM: choroba przerzutowa u dzieci poniżej 18 miesiąca życia, w której rak rozprzestrzenił się wyłącznie na skórę, wątrobę i/lub szpik kostny.
Komplikacje i konsekwencje
Neuroblastoma charakteryzuje się powikłaniami i konsekwencjami takimi jak:
- rozprzestrzenianie się (przerzuty) do węzłów chłonnych, szpiku kostnego, wątroby, skóry i kości;
- ucisk rdzenia kręgowego (który może powodować ból i prowadzić do paraliżu);
- rozwój zespołu paranowotworowego (w wyniku działania niektórych substancji chemicznych wydzielanych przez guz, a także antygenu disialogangliozydu GD2 wyrażanego przez jego komórki), który objawia się szybkimi, mimowolnymi ruchami gałek ocznych, zaburzeniami koordynacji, skurczami mięśni i biegunką;
- nawroty po zakończeniu leczenia podstawowego (jak pokazuje praktyka kliniczna, w przypadku neuroblastomów wysokiego ryzyka nawrót choroby występuje w 50% przypadków).
Diagnostyka nerwiakowłókniaki
Rozpoznanie podejrzenia neuroblastomy u dziecka wymaga przeprowadzenia badania, testów laboratoryjnych i obrazowania.
Wykonuje się badania krwi i moczu w celu wykrycia katecholamin (noradrenaliny i dopaminy) oraz kwasów homowaniliowego lub wanilinomigdałowego (powstających podczas metabolizmu tych hormonów); badanie krwi w celu wykrycia neurospecyficznej enolazy, test immunoenzymatyczny (ELISA) surowicy krwi oraz analizę szpiku kostnego (którego próbkę pobiera się przez nakłucie aspiracyjne). Wykonuje się test DNA w celu określenia mutacji, a także biopsję w celu badania cytomorfologicznego tkanki guza.
Po pobraniu próbek biopsji są one wysyłane do laboratorium, gdzie patolog (lekarz, który ma specjalne przeszkolenie w zakresie identyfikacji komórek nowotworowych) ogląda je pod mikroskopem. Często wykonuje się również specjalne testy laboratoryjne na próbkach, aby wykazać, czy guz jest neuroblastomą.
Jeśli jest to neuroblastoma, badania laboratoryjne mogą również pomóc w określeniu szybkości wzrostu lub rozprzestrzeniania się guza oraz w ustaleniu, jakie leczenie będzie najskuteczniejsze.
Diagnostyka instrumentalna pozwala na uwidocznienie nowotworu za pomocą badania USG, RTG, MRI lub TK, PET z wprowadzeniem 18F-fluorodeoksyglukozy lub skaningu MIBG - scyntygrafii z metajodobenzyloguanidyną. [ 1 ]
Diagnostyka różnicowa
Diagnostyka różnicowa obejmuje łagodnego ganglioneuroma, ganglioneuroblastoma, mięsaka prążkowanokomórkowego, nerczaka zarodkowego.
Z kim się skontaktować?
Leczenie nerwiakowłókniaki
W przypadku neuroblastomy leczenie zależy od grupy ryzyka pacjenta (stadium procesu nowotworowego), lokalizacji guza, cech genomicznych komórek nowotworowych i wieku dziecka. Może ono obejmować monitorowanie, operację, chemioterapię, radioterapię, immunoterapię i przeszczep komórek macierzystych układu krwiotwórczego.
Neoadjuwantowa lub adiuwantowa (przed- lub pooperacyjna) chemioterapia neuroblastoma u dzieci, podobnie jak każda chemioterapia na raka, jest podawana w cyklach: lek jest podawany przez kilka dni z rzędu, po czym następuje przerwa, aby organizm mógł się zregenerować. Cykle są zwykle powtarzane co trzy do czterech tygodni.
Stosuje się następujące leki (oraz ich połączenia): cyklofosfamid, cisplatyna lub karboplatyna, doksorubicyna (adriamycyna), winkrystyna, etopozyd.
Częste skutki uboczne leków chemioterapeutycznych obejmują wypadanie włosów, utratę apetytu, zmęczenie, nudności i wymioty, owrzodzenia jamy ustnej, biegunkę lub zaparcia. Chemioterapia może uszkodzić szpik kostny i spowodować zmniejszenie liczby krwinek.
Immunoterapia ukierunkowana (skierowana na antygen guza GD2) wykorzystuje leki z grupy przeciwciał monoklonalnych (anty-GD2 MAb) Dinutuximab (Unituxin) i Naxitamab. Są one podawane dożylnie w przedłużonym wlewie, w połączeniu z czynnikiem stymulującym kolonie granulocytów i makrofagów (cytokina GM-CSF) i interleukiną-2.
Do skutków ubocznych tych leków należą: ból (często bardzo silny), obniżone ciśnienie krwi, przyspieszone bicie serca, duszność (z możliwym obrzękiem dróg oddechowych), podwyższona temperatura, nudności, wymioty i biegunka, zmiany w składzie komórkowym i mineralnym krwi.
Aby zmniejszyć ryzyko nawrotu nowotworu po chemioterapii wysokodawkowej i przeszczepie komórek macierzystych, u dzieci z neuroblastomą wysokiego ryzyka stosuje się retinoidy ogólnoustrojowe – kwas 13-cis-retinowy (izotretynoina). [ 2 ]
Leczenie chirurgiczne neuroblastoma – usunięcie guza, np. otwarta adrenalektomia lub laparoskopowa resekcja neuroblastoma nadnerczy; limfektomia (usunięcie zajętych węzłów chłonnych) itp. [ 3 ]
W przypadku neuroblastomy wysokiego ryzyka można zastosować radioterapię.[4 ]
Zapobieganie
Biorąc pod uwagę przyczyny neuroblastomy u dzieci, jedynym środkiem zapobiegawczym może być poradnictwo genetyczne przy planowaniu ciąży. Należy jednak pamiętać, że ten guz jest związany z mutacjami dziedzicznymi tylko w 1-2% przypadków.
Prognoza
Neuroblastoma wieku dziecięcego ma zdolność do samoistnej regresji.
Markery prognostyczne
- Nowotwory wysokiego ryzyka, a także neuroblastoma u dzieci w każdym wieku i na wszystkich etapach (z wyjątkiem etapu 4S) – ze zwiększoną ekspresją genu N-MYC i amplifikacją onkogenu N-Myc – mają niekorzystne rokowanie, wpływające na długość życia.
- Posiadanie komórek nowotworowych, którym brakuje pewnych części chromosomów 1 lub 11 (znanych jako delecje 1p lub 11q) wiąże się z gorszymi rokowaniami. Posiadanie dodatkowej części chromosomu 17 (zysk 17q) również wiąże się z gorszymi rokowaniami.
- Komórki neuroblastoma zawierające dużą ilość DNA dają lepsze rokowanie, zwłaszcza u dzieci poniżej 2 roku życia.
- Neuroblastoma posiadające więcej receptorów neurotrofin, zwłaszcza receptora czynnika wzrostu nerwów TrkA, ma lepsze rokowanie.
Przeżywalność według grupy ryzyka Childhood Oncology Group (COG)
- Grupa niskiego ryzyka: Dzieci z grupy niskiego ryzyka mają 5-letni wskaźnik przeżycia większy niż 95%.
- Grupa ryzyka pośredniego: Dzieci z grupy ryzyka pośredniego mają 5-letni wskaźnik przeżycia na poziomie 90–95%.
- Grupa wysokiego ryzyka: Dzieci z grupy wysokiego ryzyka mają 5-letni wskaźnik przeżycia na poziomie około 50%.
Około 15% zgonów z powodu nowotworów u dzieci jest spowodowanych przez neuroblastomę. Szanse na długoterminowe przeżycie w przypadku tego nowotworu złośliwego wysokiego ryzyka nie przekraczają 40%. Całkowity pięcioletni wskaźnik przeżycia wynosi 67-74%, 43% w grupie wiekowej od jednego do czterech lat i ponad 80% w przypadku neuroblastomy zdiagnozowanej w pierwszym roku życia.
Использованная литература