Ekspert medyczny artykułu

Nowe publikacje
Choroba Charcota-Marie-Tootha.
Ostatnia recenzja: 12.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Zanik mięśni strzałkowych, zespół Charcota-Mariego-Tootha lub choroba Charcota-Mariego-Tootha to grupa przewlekłych chorób dziedzicznych, w których dochodzi do uszkodzenia nerwów obwodowych.
Według ICD-10 w sekcji dotyczącej chorób układu nerwowego kod tej choroby to G60.0 (dziedziczna neuropatia ruchowo-czuciowa). Znajduje się ona również na liście chorób sierocych.
Epidemiologia
Według statystyk klinicznych częstość występowania wszystkich typów choroby Charcota-Mariego-Tootha na 100 tys. ludności wynosi 19 przypadków (według innych źródeł jeden przypadek na 2,5–10 tys. ludności).
CMT typu 1 stanowi około dwie trzecie przypadków (jeden przypadek na 5000 do 7000 populacji), a prawie 70% z nich jest związanych z duplikacją genu PMP22. Ponad 1,2 miliona ludzi na całym świecie cierpi na ten typ choroby.
Szacuje się, że częstość występowania CMT typu 4 wynosi 1–5 przypadków na 10 000 dzieci. [ 1 ]
Przyczyny Choroba Charcota-Marie-Tootha
Zgodnie z klasyfikacją zespołów polineuropatycznych zanik mięśni strzałkowych, zanik nerwowy Charcota-Mariego-Tootha lub choroba Charcota-Mariego-Tootha (w skrócie CMT) to genetycznie uwarunkowane polineuropatie czuciowo-ruchowe. [ 2 ]
Czyli przyczynami jego występowania są mutacje genetyczne. A w zależności od charakteru odchyleń genetycznych rozróżnia się główne typy lub rodzaje tego zespołu: demielinizacyjny i aksonalny. Do pierwszej grupy zalicza się chorobę Charcota-Mariego-Tootha typu 1 (CMT1), która występuje z powodu duplikacji genu PMP22 na chromosomie 17, który koduje transbłonowe białko mieliny obwodowej 22. W rezultacie dochodzi do segmentowej demielinizacji osłonki aksonu (wypustek komórek nerwowych) i spadku szybkości przewodzenia sygnału nerwowego. Ponadto mutacje mogą występować w niektórych innych genach.
Forma aksonalna to choroba Charcota-Mariego-Tootha typu 2 (CMT2), która atakuje same aksony i jest związana z patologicznymi zmianami w genie MFN2 w locus 1p36.22, kodującym białko błonowe mitofuzynę-2, które jest niezbędne do fuzji mitochondriów i tworzenia funkcjonalnych sieci mitochondrialnych wewnątrz komórek nerwów obwodowych. Istnieje ponad tuzin podtypów CMT2 (z mutacjami w określonych genach).
Należy zauważyć, że obecnie zidentyfikowano ponad sto genów, których uszkodzenie, przekazywane drogą dziedziczenia, powoduje różne podtypy choroby Charcota-Mariego-Tootha. Na przykład mutacje w genie RAB7 prowadzą do CMT typu 2B; zmiana genu SH3TC2 (kodującego jedno z białek błonowych komórek Schwanna) powoduje CMT typu 4C, który ujawnia się w dzieciństwie i charakteryzuje się demielinizacją neuronów ruchowych i czuciowych (istnieje kilkanaście i pół postaci typu 4 tej choroby).
Rzadka CMT typu 3 (nazywana zespołem Dejerine'a-Sottasa) zaczyna się rozwijać we wczesnym dzieciństwie i jest spowodowana mutacjami w genach PMP22, MPZ, EGR2 i innych.
W przypadku CMT typu 5 występującego w wieku 5-12 lat obserwuje się nie tylko neuropatię ruchową (w postaci niedowładu spastycznego kończyn dolnych), ale także uszkodzenie nerwów wzrokowego i słuchowego.
Osłabienie mięśni i zanik nerwu wzrokowego (z utratą wzroku), a także problemy z równowagą są typowe dla typu 6 CMT. Z kolei w typie 7 choroby Charcota-Mariego-Tootha występuje nie tylko neuropatia czuciowo-ruchowa, ale także choroba siatkówki w postaci retinitis pigmentosa.
CMT sprzężona z chromosomem X, czyli choroba Charcota-Mariego-Tootha z tetraparezą kończyn (osłabienie ruchomości obu rąk i nóg), występuje częściej u mężczyzn. Jest to typ demielinizacyjny i uważa się, że wynika z mutacji w genie GJB1 na długim ramieniu chromosomu X, który koduje koneksynę 32, białko błonowe komórek Schwanna i oligodendrocytów, regulujące przekazywanie sygnałów nerwowych. [ 3 ]
Czynniki ryzyka
Głównym czynnikiem ryzyka wystąpienia CMT jest historia rodzinna choroby, tzn. występowanie jej u bliskich krewnych.
Genetycy szacują, że jeśli oboje rodzice są nosicielami genu recesywnego autosomalnego warunkującego chorobę Charcota-Mariego-Tootha, ryzyko urodzenia dziecka, u którego rozwinie się ta choroba, wynosi 25%. Ryzyko, że dziecko będzie nosicielem tego genu (ale nie będzie miało żadnych objawów) szacuje się na 50%.
W przypadku dziedziczenia sprzężonego z chromosomem X (gdy zmutowany gen znajduje się na chromosomie X kobiety) istnieje 50% ryzyko, że matka przekaże gen swojemu synowi, u którego rozwinie się CMT. Choroba może nie wystąpić, gdy urodzi się dziewczynka, ale synowie córki (wnuki) mogą odziedziczyć wadliwy gen i choroba się rozwinie.
Patogeneza
W przypadku każdego typu choroby Charcota-Mariego-Tootha jej patogeneza jest spowodowana dziedziczną anomalią nerwów obwodowych: ruchowego i czuciowego.
W przypadku typu CMT demielinizacyjnego, zniszczenie lub uszkodzenie osłonki mielinowej, chroniącej aksony nerwów obwodowych, prowadzi do spowolnienia przekazywania impulsów nerwowych w obwodowym układzie nerwowym – pomiędzy mózgiem, mięśniami i narządami zmysłów.
W przypadku aksonowego typu choroby uszkodzone zostają same aksony, co negatywnie wpływa na siłę sygnałów nerwowych, która jest niewystarczająca do pełnej stymulacji mięśni i narządów zmysłów.
Przeczytaj także:
Jak przekazywany jest zespół Charcota-Mariego-Tootha? Wadliwe geny mogą być dziedziczone w sposób autosomalny dominujący lub autosomalny recesywny.
Najczęstszy typ, dziedziczenie autosomalne dominujące, występuje, gdy istnieje jedna kopia zmutowanego genu (przenoszona przez jednego z rodziców). Prawdopodobieństwo przekazania CMT każdemu urodzonemu dziecku szacuje się na 50%. [ 4 ]
W przypadku dziedziczenia autosomalnego recesywnego, aby rozwinęła się choroba, konieczne są dwie kopie wadliwego genu (po jednej od każdego rodzica, u którego nie występują objawy choroby).
W 40-50% przypadków występuje demielinizacja dziedziczna autosomalna dominująca, czyli CMT typu 1; w 12-26% przypadków CMT aksonowa, czyli typ 2. W 10-15% przypadków obserwuje się dziedziczenie sprzężone z chromosomem X. [ 5 ]
Objawy Choroba Charcota-Marie-Tootha
Zazwyczaj pierwsze objawy tej choroby zaczynają pojawiać się w dzieciństwie i okresie dojrzewania i stopniowo rozwijają się przez całe życie, chociaż zespół ten może ujawnić się później. Połączenie objawów jest zmienne, a tempo postępu choroby, jak również jej nasilenie, jest niemożliwe do przewidzenia.
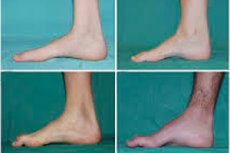
Typowe objawy początkowego stadium obejmują zwiększone ogólne zmęczenie; obniżone napięcie (osłabienie) mięśni stóp, kostek i piszczeli; brak odruchów. Utrudnia to ruchy stóp i prowadzi do dysbazji (zaburzeń chodu) w postaci wyższego unoszenia nóg, często z częstymi potknięciami i upadkami. Objawy choroby Charcota-Mariego-Tootha u małego dziecka mogą obejmować wyraźną niezdarność i nietypowe dla wieku trudności w chodzeniu związane z obustronnym opadaniem stóp. Charakterystyczne są również deformacje stóp: wysokie sklepienie (stopa pusta) lub poważne płaskostopie, zakrzywione (młotkowate) palce.
W przypadku chodzenia na palcach przy jednoczesnym niedociśnieniu mięśniowym neurolog może podejrzewać, że dziecko cierpi na CMT typu 4, charakteryzującą się tym, że dzieci mogą nie być w stanie chodzić już w okresie dojrzewania.
W miarę postępu choroby zanik mięśni i osłabienie rozprzestrzeniają się na kończyny górne, co utrudnia wykonywanie drobnych czynności motorycznych i normalnych czynności ręcznych. Zmniejszone wrażenia dotykowe i zdolność odczuwania ciepła i zimna, a także drętwienie stóp i dłoni wskazują na uszkodzenie aksonów nerwów czuciowych.
W chorobie Charcota-Mariego-Tootha typu 3 i 6, która ujawnia się w dzieciństwie, obserwuje się ataksję sensoryczną (zaburzenia koordynacji ruchów i równowagi), drżenie mięśni, uszkodzenie nerwu twarzowego, zanik nerwu wzrokowego z oczopląsem oraz utratę słuchu.
W późniejszych stadiach mogą pojawić się niekontrolowane drżenia (drżenie) i częste skurcze mięśni; trudności w poruszaniu się mogą prowadzić do rozwoju bólu: mięśniowego, stawowego, neuropatycznego.
Komplikacje i konsekwencje
Choroba Charcota-Mariego-Tootha może powodować powikłania i konsekwencje takie jak:
- częstsze skręcenia i złamania;
- przykurcze związane ze skróceniem mięśni i ścięgien okołostawowych;
- skolioza (skrzywienie kręgosłupa);
- problemy z oddychaniem – gdy uszkodzone są włókna nerwowe unerwiające mięśnie przepony:
- utrata zdolności samodzielnego poruszania się.
Diagnostyka Choroba Charcota-Marie-Tootha
Diagnozę stawia się na podstawie badania klinicznego, wywiadu (łącznie z wywiadem rodzinnym), badania neurologicznego i ogólnego.
Testy wykonuje się w celu sprawdzenia zakresu ruchu, wrażliwości i odruchów ścięgnistych. Przewodnictwo nerwowe można ocenić za pomocą diagnostyki instrumentalnej – elektromiografii lub elektroneuromiografii. Może być również wymagane badanie USG lub MRI. [ 6 ]
Testy genetyczne lub DNA w celu wykrycia najczęstszych mutacji genetycznych powodujących CMT w próbce krwi są ograniczone, ponieważ testy DNA nie są obecnie dostępne dla wszystkich typów choroby. Aby uzyskać więcej informacji, zobacz Testy genetyczne
W niektórych przypadkach wykonuje się biopsję nerwu obwodowego (najczęściej nerwu łydkowego).
Diagnostyka różnicowa
Diagnostyka różnicowa obejmuje inne neuropatie obwodowe, dystrofię mięśniową Duchenne'a, zespoły mielopatyczne i miasteniczne, neuropatię cukrzycową, mielopatie w stwardnieniu rozsianym i stwardnieniu zanikowym bocznym, zespół Guillaina-Barrégo, uraz nerwu strzałkowego i jego zanik (w tym w przypadku ucisku między dyskami lędźwiowymi kręgosłupa), uszkodzenie móżdżku lub wzgórza, a także skutki uboczne chemioterapii (podczas leczenia cytostatykami, takimi jak winkrystyna lub paklitaksel). [ 7 ]
Z kim się skontaktować?
Leczenie Choroba Charcota-Marie-Tootha
Obecnie leczenie tej dziedzicznej choroby obejmuje terapię ruchową (mającą na celu wzmocnienie i rozciąganie mięśni), terapię zajęciową (która pomaga pacjentom z osłabieniem mięśni rąk) oraz stosowanie urządzeń ortopedycznych ułatwiających chodzenie. W razie potrzeby przepisuje się środki przeciwbólowe lub przeciwdrgawkowe. [ 8 ]
W przypadkach skrajnego płaskostopia można wykonać osteotomię, natomiast w przypadku deformacji pięty wskazana jest korekta chirurgiczna – artrodeza. [ 9 ]
Trwają badania zarówno nad genetycznym składnikiem choroby, jak i nad metodami jej leczenia. Stosowanie komórek macierzystych, niektórych hormonów, lecytyny lub kwasu askorbinowego nie przyniosło jeszcze pozytywnych rezultatów.
Ale dzięki niedawnym badaniom coś nowego może rzeczywiście pojawić się w leczeniu choroby Charcota-Mariego-Tootha w niedalekiej przyszłości. Tak więc od 2014 roku francuska firma Pharnext opracowuje, a od połowy 2019 roku trwają badania kliniczne leku PXT3003 do leczenia CMT typu 1 u dorosłych, który hamuje zwiększoną ekspresję genu PMP22, poprawia mielinizację nerwów obwodowych i łagodzi objawy nerwowo-mięśniowe.
Specjaliści z firmy medycznej Sarepta Therapeutics (USA) pracują nad stworzeniem terapii genowej na chorobę Charcota-Mariego-Tootha typu 1. Terapia ta będzie wykorzystywać nieszkodliwy wirus adenowirusowy (AAV) z rodzaju Dependovirus z liniowym jednoniciowym genomem DNA, który przeniesie gen NTF3 do organizmu, kodujący białko neurotrofiny-3 (NT-3) niezbędne do funkcjonowania komórek nerwowych Schwanna.
Firma Helixmith rozpocznie do końca 2020 roku badania kliniczne opracowanej w Korei Południowej terapii genowej Engensis (VM202) w celu leczenia objawów mięśniowych w przypadku CMT typu 1. [ 10 ]
Zapobieganie
Zapobieganie CMT może polegać na poradnictwie genetycznym przyszłych rodziców, zwłaszcza jeśli ktoś w parze ma rodzinną historię choroby. Jednakże zidentyfikowano przypadki de novo punktowych mutacji genów, czyli w przypadku braku choroby w historii rodzinnej.
Prawdopodobieństwo wystąpienia choroby Charcota-Mariego-Tootha u przyszłego dziecka można sprawdzić w czasie ciąży, wykonując biopsję kosmówki (od 10. do 13. tygodnia ciąży) oraz badanie płynu owodniowego (w 15.-18. tygodniu).
Prognoza
Ogólnie rzecz biorąc, rokowanie w przypadku różnych typów choroby Charcota-Mariego-Tootha zależy od ciężkości klinicznej, ale we wszystkich przypadkach choroba postępuje powoli. Wielu pacjentów ma niepełnosprawność, chociaż nie skraca to oczekiwanej długości życia.