Ekspert medyczny artykułu

Nowe publikacje
Priony - czynniki wywołujące choroby prionowe
Ostatnia recenzja: 06.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Powolne infekcje wirusowe charakteryzują się następującymi kryteriami:
- niezwykle długi okres inkubacji (miesiące, lata);
- specyficzne uszkodzenie narządów i tkanek, przede wszystkim ośrodkowego układu nerwowego;
- powolny, stały postęp choroby;
- nieunikniony, śmiertelny skutek.

Niektóre patogeny, które powodują ostre infekcje wirusowe, mogą również powodować powolne infekcje wirusowe. Na przykład wirus odry czasami powoduje SSPE, a wirus różyczki powoduje postępującą wrodzoną różyczkę i zapalenie mózgu wywołane przez różyczkę.
Typowa powolna infekcja wirusowa u zwierząt jest powodowana przez wirus visna/madi, który jest retrowirusem. Jest czynnikiem wywołującym powolną infekcję wirusową i postępujące zapalenie płuc u owiec. Biała substancja mózgu ulega zniszczeniu, rozwija się paraliż (visna - zanik); występuje przewlekłe zapalenie płuc i śledziony.
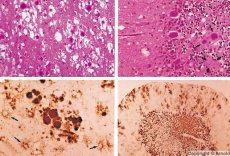
Choroby podobne w swoich cechach do powolnych infekcji wirusowych są wywoływane przez priony - czynniki wywołujące zakażenia prionowe. Choroby prionowe to grupa postępujących zaburzeń ośrodkowego układu nerwowego ludzi i zwierząt. U ludzi dochodzi do upośledzenia funkcji ośrodkowego układu nerwowego, zmian osobowości, zaburzeń ruchowych. Objawy choroby trwają zazwyczaj od kilku miesięcy do kilku lat, kończąc się śmiercią. Wcześniej zakażenia prionowe były rozpatrywane razem z tzw. czynnikami wywołującymi powolne zakażenia wirusowe.
Niektóre czynniki wywołujące choroby prionowe gromadzą się najpierw w tkankach limfoidalnych. Priony, dostając się do mózgu, gromadzą się w dużych ilościach, powodując amyloidozę (dysproteinozę pozakomórkową, charakteryzującą się odkładaniem się amyloidu z rozwojem zaniku i stwardnienia tkanki) i astrocytozę (proliferację astrocytarnych neuroglii, hiperprodukcję włókien glejowych). Tworzą się fibryle, agregaty białka lub amyloidu oraz zmiany gąbczaste w mózgu (przenośnych encefalopatii gąbczastych). W wyniku tego dochodzi do zmian zachowania, upośledzenia koordynacji ruchów, wyczerpania ze skutkiem śmiertelnym. Nie wytwarza się odporność. Choroby prionowe to choroby konformacyjne, które rozwijają się w wyniku nieprawidłowego zwinięcia (naruszenia prawidłowej konformacji) białka komórkowego niezbędnego do prawidłowego funkcjonowania organizmu. Drogi transmisji prionów są różne:
- droga pokarmowa - zakażone produkty pochodzenia zwierzęcego, dodatki do żywności z surowych narządów wołowych itp.:
- przeniesienie zakażenia poprzez transfuzję krwi, podawanie leków pochodzenia zwierzęcego, przeszczepianie narządów i tkanek, stosowanie zakażonych narzędzi chirurgicznych i stomatologicznych;
- przeniesienie poprzez preparaty immunobiologiczne (znane jest zakażenie 1500 owiec wirusem PrP''' szczepionką zawierającą preparaty mózgowo-formolowe od chorych owiec).
Patologiczne priony, po przedostaniu się do jelita, są transportowane do krwi i limfy. Po obwodowej replikacji w śledzionie, wyrostku robaczkowym, migdałkach i innych tkankach limfatycznych, są przenoszone do mózgu przez nerwy obwodowe (neuroinwazja). Możliwa jest bezpośrednia penetracja prionów do mózgu przez barierę krew-mózg. Wcześniej uważano, że ośrodkowy układ nerwowy jest jedyną tkanką, w której gromadzą się patologiczne priony, ale pojawiły się badania, które zmieniły tę hipotezę. Okazało się, że gromadzenie się prionów w śledzionie jest związane ze wzrostem i funkcjonowaniem komórek dendrytycznych pęcherzykowych.
 [
[ Właściwości prionów
Normalna komórkowa izoforma białka prionowego o masie cząsteczkowej 33-35 kDa jest determinowana przez gen białka prionowego (gen prionowy - PrNP znajduje się na 20. ludzkim chromosomie). Normalny gen pojawia się na powierzchni komórki (zakotwiczony w błonie przez glikoproteinę cząsteczki), wrażliwy na proteazę. Reguluje przekazywanie impulsów nerwowych, cykle dobowe, procesy utleniania, uczestniczy w metabolizmie miedzi w ośrodkowym układzie nerwowym i w regulacji podziału komórek macierzystych szpiku kostnego. Ponadto gen prionowy występuje w śledzionie, węzłach chłonnych, skórze, przewodzie pokarmowym i komórkach dendrytycznych pęcherzyków.
Proliferacja patologicznych prionów
Przekształcenie prionów w zmienione formy następuje, gdy równowaga kinetycznie kontrolowana między nimi zostaje zakłócona. Proces ten jest wzmacniany przez zwiększenie ilości prionów patologicznych (PrP) lub egzogennych. PrP jest normalnym białkiem zakotwiczonym w błonie komórkowej. PrP' jest globularnym hydrofobowym białkiem, które tworzy agregaty z samym sobą i PrP'' na powierzchni komórki: w rezultacie PrP' jest przekształcane w PrP'', a następnie cykl jest kontynuowany. Patologiczna forma PrP''' gromadzi się w neuronach, nadając komórce gąbczasty wygląd.
Kuru
Choroba prionowa, wcześniej powszechna wśród Papuasów (oznaczająca drżenie lub trzęsienie się) we wschodniej części wyspy Nowa Gwinea. Zakaźne właściwości choroby udowodnił K. Gajdusek. Patogen przenoszony jest drogą pokarmową w wyniku rytualnego kanibalizmu - zjadania niedogotowanego, zainfekowanego prionami mózgu zmarłych krewnych. W wyniku uszkodzenia ośrodkowego układu nerwowego upośledzone są ruchy i chód, pojawiają się dreszcze i euforia („śmiejąca się śmierć”). Okres inkubacji trwa 5-30 lat. Chory umiera po roku.
Choroba Creutzfeldta-Jakoba
Choroba prionowa, która objawia się demencją, zaburzeniami wzroku i móżdżku oraz zaburzeniami ruchu ze skutkiem śmiertelnym po 4-5 miesiącach choroby w klasycznej odmianie choroby Creutzfeldta-Jakoba i po (3-14 miesiącach w nowej odmianie choroby Creutzfeldta-Jakoba. Okres inkubacji może sięgać 20 lat. Możliwe są różne drogi zakażenia i przyczyny choroby:
- w przypadku spożywania niewystarczająco poddanych obróbce cieplnej produktów zwierzęcych, takich jak mięso i mózgi krów chorych na gąbczastą encefalopatię bydła;
- podczas przeszczepiania tkanek, np. przeszczepiania rogówki, transfuzji krwi, stosowania hormonów i innych substancji biologicznie czynnych pochodzenia zwierzęcego, stosowania katgutu, zanieczyszczonych lub niedostatecznie wysterylizowanych narzędzi chirurgicznych, manipulacji prosektoryjnych;
- w przypadku hiperprodukcji PrR i innych stanów, które stymulują proces przekształcania PrR' w PrR".
Choroba może również rozwinąć się w wyniku mutacji lub insercji w regionie genu prionowego. Rodzinny charakter choroby jest powszechny ze względu na genetyczną predyspozycję do choroby Creutzfeldta-Jakoba. W nowym wariancie choroby Creutzfeldta-Jakoba zaburzenia rozwijają się w młodszym wieku (średni wiek 28 lat), w przeciwieństwie do klasycznego wariantu (średni wiek 65 lat). W nowym wariancie choroby Creutzfeldta-Jakoba nieprawidłowe białko prionowe gromadzi się nie tylko w ośrodkowym układzie nerwowym, ale także w tkankach limforetikularnych, w tym migdałkach.
Zespół Gerstmanna-Sträusslera-Scheinkera
Dziedziczna choroba prionowa, której towarzyszą demencja, hipotonia, zaburzenia połykania (dysfagia), dyzartria. Często ma charakter rodzinny. Okres inkubacji wynosi od 5 do 30 lat. Choroba występuje w wieku 50-60 lat, jej czas trwania waha się od 5 do 13 lat.
Dziedziczna śmiertelna bezsenność
Choroba autoimmunologiczna z postępującą bezsennością, nadreaktywnością współczulną (nadciśnienie, hipertermia, nadpotliwość, tachykardia), drżeniem, ataksją, wieloklonowością, halucynacjami. Sen jest poważnie zaburzony. Śmierć następuje wraz z postępem niewydolności sercowo-naczyniowej.
Zeskrobać
Scrapie (od angielskiego scrape – drapać) jest chorobą prionową owiec i kóz (świerzb), która objawia się uszkodzeniem ośrodkowego układu nerwowego, postępującymi zaburzeniami ruchu, silnym świądem skóry (świerzb) i kończy się śmiercią zwierzęcia.
Gąbczasta encefalopatia bydła
Choroba bydła charakteryzująca się uszkodzeniem ośrodkowego układu nerwowego, upośledzeniem koordynacji ruchów i nieuchronną śmiercią zwierzęcia. Epidemia choroby wybuchła najpierw w Wielkiej Brytanii. Związana była z karmieniem zwierząt mączką mięsno-kostną zawierającą patologiczne priony. Okres inkubacji wynosi od 1,5 do 15 lat. Najbardziej zakażeniu ulegają mózg, rdzeń kręgowy i gałki oczne zwierząt.
Diagnostyka laboratoryjna chorób prionowych
Podczas diagnostyki stwierdza się zmiany gąbczaste w mózgu, astrocytozę (gliozę) i brak nacieków zapalnych. Mózg jest barwiony na amyloid. W płynie mózgowo-rdzeniowym wykrywa się markery białkowe zaburzeń mózgu wywołanych prionami (za pomocą testu ELISA). Przeprowadza się analizę genetyczną genu prionowego (PCR).
Zapobieganie chorobom prionowym
Autoklawowanie (w temp. 134°C przez 18 min; w temp. 121°C przez 1 h), spalanie, dodatkowe traktowanie wybielaczem i roztworem NaCl o normalnym stężeniu przez 1 h jest zalecane w celu odkażania narzędzi i przedmiotów środowiskowych. W profilaktyce niespecyficznej wprowadzono ograniczenia dotyczące stosowania produktów leczniczych pochodzenia zwierzęcego, a produkcja hormonów przysadkowych pochodzenia zwierzęcego jest zabroniona. Ograniczono przeszczepianie opony twardej. Podczas pracy z płynami dialogowymi pacjentów stosuje się rękawice gumowe.