
Dziedziczne zapalenie nerek (zespół Alporta) u dzieci
Ostatnia recenzja: 23.04.2024

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Dziedziczne nerek (zespół alporta) - uwarunkowane genetycznie nieimmunologicznych dziedziczona kłębków nerkowych wykazujących krwiomocz (czasami białkomocz), postępujące zaburzenie czynności nerek, przewlekłej niewydolności nerek rozwoju często wiąże się z odbiorczym głuchoty oraz niedowidzących.
Po raz pierwszy choroba została opisana w 1902 roku przez LGGuthrie, który zaobserwował rodzinę w kilku pokoleniach, z których obserwowano krwiomocz. W 1915 r. Członkowie tej samej rodziny AFHurst opisali rozwój mocznicy. W 1927 r. A Alport po raz pierwszy zidentyfikował głuchotę u kilku krewnych z krwiomoczem.W latach 50. Ubiegłego wieku urazów oczu opisano w tej chorobie. W 1972 roku, u pacjentów z dziedziczną hematurią, morfologicznie badając tkankę nerkową, Hinglais et al. Ujawniła nierównomierne rozszerzanie i delaminację kłębuszkowych błon podstawnych. W 1985 roku zidentyfikowano genetyczną podstawę dziedzicznego zapalenia nerek - mutację w genie kolagenu typu IV (Fiengold i wsp., 1985).
Badanie genetycznej natury choroby pozwoliło na wyciągnięcie wniosku, że różnice w fenotypowych przejawach dziedzicznego zapalenia nerek (z ubytkiem słuchu lub bez) wynikają z stopnia ekspresji zmutowanego genu. Zatem obecnie wszystkie warianty kliniczne są uważane za przejawy jednej choroby, a termin "dziedziczne zapalenie nerek" jest równoznaczny z terminem "zespół Alport".
Według badań epidemiologicznych, dziedziczne zapalenie nerek występuje z częstością 17 na 100 000 dzieci.
Przyczyny zespołu Alporta
Podstawą genetyczną choroby jest mutacja w genie α-5 łańcucha kolagenu typu IV. Ten typ jest uniwersalny dla podstawowych błon nerkowych, ślimakowych, soczewkowych, siatkówki i rogówki oka, co udowodniono w badaniach z użyciem przeciwciał monoklonalnych przeciwko tej frakcji kolagenu. Ostatnio wskazują na możliwość zastosowania sond DNA do prenatalnej diagnostyki dziedzicznego zapalenia nerek.
Podkreślono znaczenie testowania wszystkich członków rodziny za pomocą sond DNA w celu identyfikacji nosicieli zmutowanego genu, co ma ogromne znaczenie w prowadzeniu medycznych poradnictwa genetycznego rodzin z tą chorobą. Jednak do 20% rodzin nie ma krewnych z chorobą nerek, co sugeruje dużą częstość występowania spontanicznych mutacji w nieprawidłowym genie. Większość pacjentów z dziedzicznym zapaleniem nerek w rodzinach ma osoby z chorobą nerek, utratą słuchu i patologią widzenia; pokrewne małżeństwa między osobami, które mają jednego lub więcej przodków, ponieważ małżeństwo pokrewnych osób zwiększa prawdopodobieństwo uzyskania tych samych genów od obojga rodziców. Dominuje autosomalny dominujący i autosomalny recesywny i dominujący, związany z chromosomem X szlaku transmisyjnego.
Dzieci częściej odróżniają trzy warianty dziedzicznego zapalenia nerek: zespół Alporta, dziedziczne zapalenie nerek bez utraty słuchu i łagodny krwiomocz rodzinny.
Zespół Alporta - dziedziczne zapalenie nerek z uszkodzeniem słuchu. Podstawą jest połączony defekt w strukturze kolagenu błony podstawnej kłębuszków nerkowych, struktur ucha i oka. Gen klasycznego zespołu Alporta znajduje się w locus 21-22 q długiego ramienia chromosomu X. W większości przypadków jest dziedziczony przez dominujący typ związany z chromosomem X. Pod tym względem u mężczyzn zespół Alporta jest trudniejszy, ponieważ u kobiet funkcja zmutowanego genu jest kompensowana przez zdrowy allel drugiego, nienaruszonego chromosomu.
Genetyczną podstawą rozwoju dziedzicznego zapalenia nerek są mutacje w genach łańcuchów alfa kolagenu typu IV. Wiadomo, że sześć łańcuchów A kolagenu IV typu G: geny łańcuchów a5- i a6 (Co4A5 i Co4A5) znajdują się na długim ramieniu chromosomu X w strefie 21-22q; geny łańcuchów a3 i a4 (Co4A3 i Co4A4) - na drugim chromosomie; geny łańcuchów a1 i a2 (Co4A1 i Co4A2) - na 13 chromosomie.
W większości przypadków (80-85%), związany z X rodzaj dziedziczenia choroby jest związany z uszkodzeniem genu Co4A5 z powodu delecji, mutacji punktowych lub zaburzeń splicingu. Obecnie znajduje się ponad 200 mutacji genu Kol4A5, odpowiedzialnego za naruszenie syntezy łańcuchów a5 kolagenu typu IV. W przypadku tego rodzaju dziedziczenia choroba przejawia się u dzieci obojga płci, ale u chłopców jest to trudniejsze.
Mutacje w loci genów Co4A3 i Co4A4, odpowiedzialnych za syntezę a3 i a4 - łańcuchów kolagenu typu IV, są dziedziczone autosomalnie. Według badań autosomalny dominujący typ dziedziczenia obserwuje się w 16% przypadków dziedzicznego zapalenia nerek, autosomalnie recesywnego - u 6% pacjentów. Istnieje około 10 mutacji genów Co4A3 i Co4A4.
Wynikiem mutacji jest naruszenie procesów montażu kolagenu typu IV, co prowadzi do zakłócenia jego struktury. Kolagen typu IV jest jednym z głównych składników błony podstawnej kłębuszków, aparatu ślimakowego i soczewki oka, których patologia ujawnia się w klinice dziedzicznego zapalenia nerek.
Kolagenu typu IV, część błony podstawnej kłębuszków, składa się zasadniczo z dwóch łańcuchów a1 (IV) i jeden łańcuch a2 (IV), a także zawiera a3, a4, a5 łańcucha. Najczęściej, gdy X-linked dziedziczenie Sol4A5 mutacja towarzyszy brak A3, A4, A5 i A6 łańcuchy kolagenu typu IV w strukturze, a liczba O1 i łańcuchów a2 do błony podstawnej kłębuszków nerkowych wzrasta. Mechanizm tego zjawiska jest niejasny, zakłada się, że przyczyną są posttransskrypcyjne zmiany w mRNA.
Brak A3, A4 i A5 łańcuchy w strukturach typu IV kolagenu podstawnej błony kłębuszków powoduje przerzedzenie i kruchości wczesnych etapach zespół alporta że manifestuje się klinicznie większość krwiomocz (czasami krwiomocz lub białkomocz tylko białkomocz), straty i lenticonus słuchu. Dalszej progresji choroby prowadzi do zgrubienia i zakłócenia przepuszczalności błony podstawnej w późnych stadiach choroby, ze wzrostem tych rodzajów kolagenu V i VI i przejawia się wzrostem białkomoczu i niewydolnością nerek.
Natura mutacji leżących u podstaw dziedzicznego zapalenia nerek w dużej mierze determinuje jego fenotypową manifestację. Gdy X delecji chromosomu przy jednoczesnym mutacji i Sol4A6 Sol4A5 genów odpowiedzialnych za syntezę A5 i A6 łańcuchów kolagenu typu IV, w połączeniu z zespołem Alporta leiomyomatosis przełyku i narządów płciowych. Według badań z Sol4A5 mutacji związanych z delecją są oznaczone duże nasilenie procesu patologicznego, połączenie z uszkodzeniem nerek extrarenal przejawy i wczesnego rozwoju przewlekłej niewydolności nerek, w porównaniu stochechnoy mutacje tego genu.
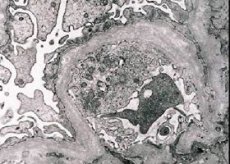
Morfologicznie mikroskopia elektronowa ujawnia rozrzedzenie i delaminację kłębuszkowych błon podstawnych (zwłaszcza blaszki densa) oraz obecność gęstych granulek elektronowych. Uszkodzenie kłębuszków może być niejednorodne u tego samego pacjenta, od minimalnej zmiany ogniskowej mezangium do stwardnienia kłębuszków nerkowych. Zapalenie kłębuszków nerkowych w zespole Alporta ma zawsze charakter immuno-ujemny, co odróżnia go od kłębuszkowego zapalenia nerek. Charakterystyczne są: zanik kanału, naciek limfohistiocytów, obecność "komórek piankowatych" z wtrętami lipidowymi - lipofagi. Wraz z postępem choroby ujawnia się pogrubienie i znaczne zniszczenie podstawowych błon kłębuszkowych.
Ujawniono pewne zmiany w stanie układu odpornościowego. U pacjentów z dziedzicznym zapaleniem nerek obserwuje się spadek poziomu IgA i tendencję do zwiększania stężenia IgM we krwi, poziom IgG można zwiększyć we wczesnych stadiach rozwoju choroby i późno obniżyć. Być może wzrost stężenia IgM i G jest rodzajem kompensacyjnej odpowiedzi w odpowiedzi na deficyt IgA.
Aktywność czynnościowa układu limfocytów T jest zmniejszona; występuje selektywne zmniejszenie liczby limfocytów B odpowiedzialnych za syntezę Ig A, naruszenie fagocytarnego odporności, głównie z powodu naruszenia chemotaksji i wewnątrzkomórkowego trawienia w neutrofilach
W badaniu biopsji nerek u pacjentów z zespołem Alport mikroskopem elektronowym ultrastrukturalne zmiany obserwowane kłębuszkowej błony podstawnej: rozcieńczenie, a naruszenie Wzory rozszczepienia kłębuszkowego błony podstawnej i zmiany jej grubości i nierównym terenie. We wczesnych stadiach dziedzicznego zapalenia nerek defekt określa rozrzedzenie i kruchość błony podstawnej kłębuszków nerkowych.
Rozrzedzenie błon kłębuszkowych jest bardziej korzystnym objawem i częściej występuje u dziewcząt. Bardziej stałą cechą mikroskopu elektronowego w dziedzicznym zapaleniu nerek jest rozszczepienie błony podstawnej, a stopień jej zniszczenia koreluje z ciężkością procesu.

Objawy zespołu Alport u dzieci
Pierwsze objawy zespołu Alporta w postaci wyizolowanego zespołu moczowego są częściej wykrywane u dzieci w pierwszych trzech latach życia. W większości przypadków choroba jest wykryta przez przypadek. Zespół moczu ujawnia się podczas badania profilaktycznego dziecka, przed wejściem do placówki dziecięcej lub podczas ARVI. W przypadku pojawienia się patologii w moczu podczas ARVI. W dziedzicznym zapaleniu nerek, w przeciwieństwie do nabytego kłębuszkowego zapalenia nerek, nie ma okresu utajonego.
W początkowej fazie choroby dobrostan dziecka cierpi na niedosyt, cechą charakterystyczną jest uporczywość i trwałość zespołu moczowego. Jednym z głównych objawów jest krwiomocz o różnym nasileniu, obserwowany w 100% przypadków. Wzrost stopnia krwiomoczu obserwuje się podczas lub po zakażeniach dróg oddechowych, wysiłku fizycznym lub po szczepieniach zapobiegawczych. Białkomocz w większości przypadków nie przekracza 1 g / dobę, na początku choroby może być niestabilny, w miarę postępu procesu wzrasta białkomocz. Okresowo osadu moczu może mieć leukocyturię z przewagą limfocytów, co jest związane z rozwojem zmian śródmiąższowych.
Później dochodzi do naruszenia częściowych funkcji nerek, pogorszenia stanu ogólnego pacjenta: zatrucia, osłabienia mięśni, niedociśnienia tętniczego, często upośledzenia słuchu (szczególnie u chłopców), czasem upośledzenia wzroku. Zatrucie objawia się bladością, zmęczeniem, bólami głowy. W początkowej fazie choroby utrata słuchu w większości przypadków jest wykrywana tylko przez audiografię. Utrata słuchu w zespole Alport może wystąpić w różnych okresach dzieciństwa, ale najczęściej ubytek słuchu rozpoznaje się w wieku 6-10 lat. Utrata słuchu u dzieci rozpoczyna się przy wysokich częstotliwościach, osiągając znaczny stopień przewodzenia powietrza i kości, przechodząc od przenoszenia dźwięku do utraty słuchu z dźwiękiem. Utrata słuchu może być jednym z pierwszych objawów choroby i może poprzedzać zespół moczowy.
W 20% przypadków pacjenci z zespołem Alporta mają zmiany w oczach. Najczęstsze anomalie z soczewki: spherofokiya, lentikonus przedni, tylny lub mieszany, różnorodność zaćmy. W rodzinach z zespołem Alportu występuje znaczna częstość występowania krótkowzroczności. Wielu badaczy stale odnotowuje w tych rodzinach obustronne, perymistyczne zmiany w postaci jasnych białawych lub żółtawych granulek w obszarze żółtego ciała. Uważają ten objaw za stały objaw, który ma wysoką wartość diagnostyczną w zespole Alport. C. S. Chugh i in. (1993) dla okulistyczną badania wykazały pacjentów zespół alporta zmniejszenie ostrości widzenia w 66,7% przypadków przodu lenticonus - 37,8%, przy czym plamki na siatkówce - w 22,2%, zaćmę - 20%, stożek rogówki - 6 7%.
U niektórych dzieci z dziedzicznym zapaleniem nerek, zwłaszcza w przebiegu niewydolności nerek, obserwuje się znaczne opóźnienie w rozwoju fizycznym. W miarę postępu niewydolności nerek rozwija się nadciśnienie. U dzieci częściej wykrywa się go w okresie dojrzewania i w starszych grupach wiekowych.
Charakterystyczna jest obecność u pacjentów z dziedzicznym zapaleniem nerek różnego rodzaju (ponad 5-7) znamion dystygoterapii tkanki łącznej. Wśród tkanki łącznej piętno u pacjentów z najczęstszą hiperteloryzm oka, wysokiej podniebienia, wady zgryzu, zaburzenia kształtu uszu, krzywizna małego palca na rękach, „luka sandalevidnaya” na nogi. Dziedzicznych zapalenie charakteryzuje się równomierność dizembriogeneza napiętnowania obrębie rodziny, jak również wysokiej częstotliwości ich dystrybucji u badanych rodzin, dzięki którym choroba jest transmitowana.
We wczesnym stadium choroby wykazała pojedyncze zmniejszenie częściowego nerek: transport aminokwasów, elektrolitów, funkcji stężenia Acidogenesis dalsze zmiany są stan funkcjonalny zarówno w proksymalnym i dystalnym narząd i mają postać połączonych zaburzeń częściowych. Obniżenie filtracji kłębuszkowej występuje później, częściej w okresie młodzieńczym. Wraz z postępem dziedzicznego zapalenia nerek rozwija się niedokrwistość.
Tak więc, dla dziedzicznej zapalenie nerek znamienny zaawansowania choroby: pierwszego etapu lub utajone ukryte objawów klinicznych objawiających pęcherzyka zespołu minimalnymi zmianami potem następuje stopniowy proces dekompensacji ze zmniejszeniem czynności nerek jawnych objawów klinicznych (zatrucie, osłabienie, opóźnienia rozwoju, anemizatsiya). Objawy kliniczne pojawiają się zwykle niezależnie od stratyfikacji reakcji zapalnej.
Wrodzone zapalenie nerek może manifestować się w różnych okresach wiekowych, które zależą od działania genu, który do pewnego czasu znajduje się w stanie represji.
Klasyfikacja
Istnieją trzy warianty dziedzicznego zapalenia nerek
- Wariant I przejawia się klinicznie przez zapalenie nerek z krwiomoczem, utratą słuchu i uszkodzeniem oczu. Przebieg zapalenia nerek postępuje wraz z rozwojem CRF. Rodzaje dziedziczenia są dominujące, powiązane z chromosomem X. Morfologicznie występuje zaburzenie struktury błony podstawnej, jej przerzedzenie i rozszczepienie.
- Wariant II - klinicznie manifestuje się zapaleniem nerek z krwiomoczem bez utraty słuchu. Przebieg zapalenia nerek postępuje wraz z rozwojem przewlekłej niewydolności nerek. Rodzaje dziedziczenia są dominujące, powiązane z chromosomem X. Morfologicznie ujawnia się trzebież błony podstawnej kłębuszkowych naczyń włosowatych (zwłaszcza laminadensa).
- III opcja - łagodny krwiomocz krwi. Kurs jest korzystny, przewlekła niewydolność nerek nie rozwija się. Typ dziedziczenia jest autosomalny dominujący lub autosomalny recesywny. W dziedziczeniu autosomalnym recesywnym kobiety mają cięższy przebieg choroby.
Diagnoza zespołu Alporta
Proponuje się następujące kryteria:
- obecność w każdej rodzinie co najmniej dwóch pacjentów z nefropatią;
- krwiomocz jako główny objaw nefropatii w probandzie;
- co najmniej jeden członek rodziny ma ubytek słuchu;
- rozwój przewlekłej niewydolności nerek u jednego krewnego i więcej.
W diagnostyce wielu chorób dziedzicznych i wrodzonych ważnym miejscem należący do zintegrowanego podejścia do kontroli, a przede wszystkim zwrócić uwagę na danych uzyskanych w przygotowaniu rodowodzie dziecka. Diagnoza zespół alporta uważanych za ważne w przypadku, gdy pacjent 3 z 4 typowych cech: obecność w krwiomocz rodziny i przewlekłą niewydolność nerek, obecność czuciowo słuchu pacjenta, patologii wykrywania w mikroskopii elektronowej charakterystyki biopsji objawów ROZKŁADU kłębuszkowej błony podstawnej do zmiany jej grubości i nierówne kontury.
Badanie pacjenta powinno obejmować kliniczne metody genetyczne; ukierunkowane badanie anamnezy choroby; ogólne badanie pacjenta z uwzględnieniem kryteriów diagnostycznych. Na etapie kompensacji można złapać patologię jedynie poprzez skupienie się na takich zespołach, jak obecność dziedzicznej powikłań, niedociśnienie, liczne stygmatyzacje dyzaminogenezy, zmiany w zespole moczowym. W wyrównaną estrarenalnyh może wywoływać objawy zatrucia, takie jak ciężka, osłabienie, opóźnienie rozwoju fizycznego anemizatsiya przejawia i amplifikacji ze stopniowym zmniejszeniem czynności nerek. U większości pacjentów ze zmniejszeniem czynności nerek obserwuje się zmniejszenie funkcji kwaso- i aminogenezy; u 50% pacjentów obserwuje się znaczny spadek wydzielniczej funkcji nerek; ograniczenie zakresu fluktuacji gęstości optycznej moczu; naruszenie rytmu filtracji, a następnie zmniejszenie filtracji kłębuszkowej. Stopień przewlekłej niewydolności nerek rozpoznaje się u pacjentów z 3-6 miesięcznym i podwyższonym poziomem mocznika w surowicy krwi (ponad 0,35 g / l), a spadek przesączania kłębuszkowego do 25% normy.
Diagnostyka różnicowa dziedzicznego zapalenia nerek muszą być wykonywane głównie z nabytej forma hematuric kłębuszkowe zapalenie nerek. Zyskał bardziej ostre zapalenie kłębuszków nerkowych, począwszy okres 2-3 tygodni po poprzedniej infekcji extrarenal funkcje, w tym nadciśnienia z pierwszych dni (w dziedzicznej nerek odwrotnie, niedociśnienie), zmniejszenie szybkości przesączania kłębuszkowego po wystąpieniu, nie naruszenie cząstkowych funkcji rurowych, przy czym tak jak z dziedziczną są obecni. Nabyte kłębuszkowe zapalenie nerek występuje z cięższą krwiomocz i białkomocz ze zwiększoną ESR. Wartość diagnostyczna to typowe zmiany w błonie podstawnej kłębuszków nerkowych, zapalenie nerek cecha dziedziczna.
Diagnostyka różnicowa nefropatii dysmetabolicznej prowadzona jest z przewlekłą niewydolnością nerek, rodzina klinicznie identyfikuje wiele typów chorób nerek i być może spektrum nefropatii od odmiedniczkowego zapalenia nerek po kamicę moczową. Dzieci często skarżą się na bóle brzucha i okresowo z oddawaniem moczu, w osadzie z moczem - szczawian.
Jeśli podejrzewasz, że pacjent z dziedzicznym zapaleniem nerek powinien zostać wysłany w celu wyjaśnienia diagnozy w specjalistycznym wydziale nefrologii.
Co trzeba zbadać?
Jak zbadać?
Jakie testy są potrzebne?
Z kim się skontaktować?
Leczenie zespołu Alporta
W reżimie przewiduje ograniczenie dużego wysiłku fizycznego, pozostać na świeżym powietrzu. Dieta jest wysokiej jakości, z wystarczającą zawartością wysokowartościowych białek, tłuszczów i węglowodanów, biorąc pod uwagę funkcję nerek. Ogromne znaczenie ma identyfikacja i rehabilitacja przewlekłych ognisk infekcji. Od narkotyków, ATP, kokaroksylazy, pirydoksyny (do 50 mg / dzień), chlorku karnityny. Kursy odbywają się 2-3 razy w roku. Kiedy hematuria jest przepisana fitoterapią - pokrzywa, pokrzywa, popiół jeżynowy, krwawnik pospolity.
W literaturze zagranicznej i krajowej doniesiono o leczeniu prednizolonem i stosowaniu cytostatyków. Jednak efekt jest trudny do osądzenia.
W przewlekłej niewydolności nerek stosuje się hemodializę i przeszczep nerki.
Nie istnieją metody swoistej (skutecznej patogenetycznej) terapii dziedzicznego zapalenia nerek. Wszystkie środki medyczne mają na celu zapobieganie i spowalnianie zmniejszenia czynności nerek.
Dieta powinna być zbilansowana i wysokokaloryczna, biorąc pod uwagę stan funkcjonalny nerek. W przypadku braku naruszenia stanu funkcjonalnego w żywieniu dziecka powinna być wystarczająca zawartość białek, tłuszczów i węglowodanów. W przypadku objawów niewydolności nerek należy ograniczyć ilość białka, węglowodanów wapnia i fosforu, co opóźnia rozwój przewlekłej niewydolności nerek.
Stres fizyczny powinien być ograniczony, zaleca się dzieciom powstrzymanie się od uprawiania sportu.
Unikaj kontaktu z pacjentami zakaźnymi, zmniejsz ryzyko zachorowania na ostre infekcje dróg oddechowych. Konieczne jest oczyszczenie ognisk przewlekłego zakażenia. Nie przeprowadza się profilaktycznych szczepień u dzieci z dziedzicznym zapaleniem nerek, szczepienie jest możliwe tylko zgodnie ze wskazaniami epidemiologicznymi.
Hormonalna i immunosupresyjna terapia w dziedzicznym zapaleniu nerek jest nieskuteczna. Istnieją oznaki pewnego pozytywnego działania (spadek poziomu białkomoczu i spowolnienia progresji choroby) z długotrwałym stosowaniem cyklosporyny A i inhibitorów ACE przez wiele lat.
W leczeniu pacjentów stosujących leki poprawiające metabolizm:
- pirydoksyna - 2-3 mg / kg / dzień w 3 dawkach podzielonych przez 4 tygodnie;
- kokarboksilaza - 50 mg domięśniowo co drugi dzień, tylko 10-15 wstrzyknięć;
- ATP - 1 ml domięśniowo co drugi dzień, 10-15 iniekcji;
- Witamina A - 1000 U / rok / dzień w 1 recepcji przez 2 tygodnie;
- witamina E - 1 mg / kg / dzień w 1 recepcji przez 2 tygodnie.
Taka terapia poprawia ogólny stan pacjentów, zmniejsza dysfunkcję rurkową i jest podawana 3 razy w roku.
Jako immunomodulator można stosować lewamizol - 2 mg / kg / dobę 2-3 razy w tygodniu z przerwami pomiędzy dawkami 3-4 dni.
Dla badaczy hiperbaryczne natlenianie ma pozytywny wpływ na nasilenie krwiomoczu i dysfunkcji nerek.
Najskuteczniejszą metodą leczenia dziedzicznego zapalenia nerek jest terminowe przeszczepienie nerki. Nie ma nawrotu choroby w przeszczepie, w niewielkim odsetku przypadków (około 5%), możliwe jest rozwinięcie się zapalenia nerek w przeszczepionej nerce związanej z antygenami do błony podstawnej kłębuszków nerkowych.
Obiecującym obszarem jest diagnoza prenatalna i inżynieria genetyczna. Eksperymenty na zwierzętach wykazują wysoką skuteczność przenoszenia normalnych genów odpowiedzialnych za syntezę łańcuchów A kolagenu typu IV do tkanki nerkowej, po czym odnotowuje się syntezę prawidłowych struktur kolagenowych.
Prognoza
Prognozy dotyczące dziedzicznego zapalenia nerek są zawsze poważne.
Prognostycznie niekorzystne kryteria dla przepływu dziedzicznego zapalenia nerek to:
- płeć męska;
- wczesny rozwój przewlekłej niewydolności nerek u członków rodziny;
- białkomocz (więcej niż 1 g / dzień);
- pogrubienie kłębuszkowych błon podstawnych zgodnie z mikroskopem;
- zapalenie nerwu słuchowego;
- delecja w genie Co4A5.
Rokowanie łagodnego krwioczatu rodzinnego jest bardziej korzystne.
Использованная литература