
Choroba Charcota-Mariego-Tootha
Ostatnia recenzja: 23.11.2021

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Zanik mięśni strzałkowych, zespół czy choroba Charcota-Mariego-Tootha to cała grupa przewlekłych chorób dziedzicznych z uszkodzeniem nerwów obwodowych.
Według ICD-10 w sekcji chorób układu nerwowego kod tej choroby to G60.0 (dziedziczna neuropatia ruchowa i czuciowa). Znajduje się również na liście chorób sierocych.
Epidemiologia
Według statystyk klinicznych częstość występowania wszystkich typów choroby Charcota-Mariego-Tootha na 100 tys.ludności wynosi 19 przypadków (według innych źródeł jeden przypadek na 2,5-10 tys.ludności).
CMT typu 1 stanowi około dwóch trzecich przypadków (jeden przypadek na 5-7 tys. Populacji), a prawie 70% z nich wiąże się z duplikacją genu PMP22. Na świecie ten rodzaj choroby dotyka ponad 1,2 miliona ludzi.
Częstość CMT typu 4 szacuje się na 1-5 przypadków na 10 tys. Dzieci. [1]
Przyczyny choroba Charcota-Mariego-Tootha
Zgodnie z klasyfikacją zespołów polineuropatycznych zanik mięśni strzałkowych (strzałkowych), zanik nerwowy Charcota-Mariego-Tootha lub choroba Charcota-Mariego-Tootha (w skrócie CMT) odnosi się do genetycznie uwarunkowanych polineuropatii motoryczno-czuciowych. [2]
Oznacza to, że przyczyną jego wystąpienia są mutacje genetyczne. W zależności od natury nieprawidłowości genetycznych, główne typy lub typy tego zespołu różnią się: demielinizacyjny i aksonalny. Pierwsza grupa obejmuje chorobę Charcota-Mariego-Tootha typu 1 (CMT1), która występuje w wyniku duplikacji genu PMP22 na chromosomie 17, który koduje transbłonowe białko mieliny obwodowej 22. W rezultacie segmentalna demielinizacja pochewki aksonów (procesy komórek nerwowych) i następuje zmniejszenie szybkości przewodzenia nerwów. Sygnały. Ponadto mogą występować mutacje w niektórych innych genach.
Formą aksonalną jest choroba Charcota-Marie-Tootha typu 2 (CMT2), która atakuje same aksony i jest związana ze zmianami patologicznymi w genie MFN2 w locus 1p36.22, który koduje białko błonowe mitofuzynę-2, co jest konieczne do fuzji mitochondriów i tworzenia funkcjonalnych sieci mitochondrialnych w obrębie nerwów obwodowych komórek. Istnieje kilkanaście podtypów CMT2 (z mutacjami w określonych genach).
Należy zauważyć, że obecnie zidentyfikowano ponad sto genów, których uszkodzenie, odziedziczone, powoduje różne podtypy choroby Charcota-Mariego-Tootha. Na przykład mutacje w genie RAB7 powodują rozwój CMT typu 2B; zmiana genu SH3TC2 (który koduje jedno z białek błon komórkowych Schwanna) powoduje CMT typu 4C, która objawia się w dzieciństwie i charakteryzuje się demielinizacją neuronów ruchowych i czuciowych (półtora tuzina form typu 4 tę chorobę wyróżnia się).
Rzadki SMT typu 3 (zwany zespołem Dejerine-Sott), spowodowany mutacjami w genach PMP22, MPZ, EGR2 i innych, również zaczyna się rozwijać we wczesnym dzieciństwie.
W przypadku wystąpienia CMT typu 5 w wieku 5-12 lat obserwuje się nie tylko neuropatię ruchową (w postaci spastycznego niedowładu kończyn dolnych), ale także uszkodzenie nerwu wzrokowego i słuchowego.
Charakterystyczne dla CMT typu 6 jest osłabienie mięśni i zanik nerwu wzrokowego (z utratą wzroku), a także problemy z równowagą. W przypadku choroby Charcota-Marie-Tootha typu 7 obserwuje się nie tylko neuropatię ruchowo-czuciową, ale także chorobę siatkówki w postaci barwnikowego zwyrodnienia siatkówki.
Częściej występująca choroba SMT sprzężona z chromosomem X lub choroba Charcota-Mariego-Tootha z tetraparezą kończyn (osłabienie ruchu obu rąk i nóg) u mężczyzn jest typem demielinizacyjnym i jest uważana za wynik mutacji w genie GJB1 na długie ramię chromosomu X, które koduje koneksynę 32, transbłonowe białko komórki Schwanna i oligodendrocyty, które reguluje przekazywanie sygnałów nerwowych. [3]
Czynniki ryzyka
Głównym czynnikiem ryzyka CMT jest obecność tej choroby w wywiadzie rodzinnym, czyli u bliskich krewnych.
Według genetyków, jeśli oboje rodzice są nosicielami autosomalnego recesywnego genu choroby Charcota-Mariego-Tootha, ryzyko urodzenia dziecka, u którego rozwinie się ta choroba, wynosi 25%. A ryzyko, że dziecko będzie nosicielem tego genu (ale on sam nie będzie miał żadnych objawów) szacuje się na 50%.
W przypadku dziedziczenia sprzężonego z chromosomem X (gdy zmutowany gen znajduje się na chromosomie X kobiety), istnieje 50% ryzyko, że matka przekaże ten gen swojemu synowi i u niego rozwinie się choroba CMT. Kiedy rodzi się dziecko płci żeńskiej, choroba może nie wystąpić, ale synowie (wnuki) córki mogą odziedziczyć wadliwy gen - wraz z rozwojem choroby.
Patogeneza
W każdym typie choroby Charcota-Mariego-Tootha jej patogeneza jest spowodowana dziedziczną anomalią nerwów obwodowych: motorycznym (motorycznym) i czuciowym (czuciowym).
Jeśli typ CMT ma charakter demielinizacyjny, to zniszczenie lub uszkodzenie osłonki mielinowej, która chroni aksony nerwów obwodowych, prowadzi do spowolnienia transmisji impulsów nerwowych obwodowego układu nerwowego - między mózgiem, mięśniami i narządami zmysłów.
W aksonalnym typie choroby bezpośrednio dotknięte są aksony, co negatywnie wpływa na siłę sygnałów nerwowych, która jest niewystarczająca do pełnej stymulacji mięśni i narządów zmysłów.
Przeczytaj także:
Jak rozprzestrzenia się zespół Charcota-Marie-Tootha? Wadliwe geny mogą być dziedziczone w sposób autosomalny dominujący lub autosomalny recesywny.
Najczęstsze - dziedziczenie autosomalne dominujące - występuje, gdy istnieje jedna kopia zmutowanego genu (posiadana przez jednego z rodziców). Prawdopodobieństwo przeniesienia CMT na każde z urodzonych dzieci szacuje się na 50%. [4]
W przypadku dziedziczenia autosomalnego recesywnego choroba wymaga dwóch kopii wadliwego genu (po jednej od każdego z rodziców bez objawów choroby).
W 40-50% przypadków dochodzi do autosomalnej dominującej dziedzicznej demielinizacji, czyli CMT typu 1; w 12-26% przypadków - aksonalna CMT, czyli typ 2. A w 10-15% przypadków obserwuje się dziedziczenie sprzężone z X. [5]
Objawy choroba Charcota-Mariego-Tootha
Zwykle pierwsze oznaki tej choroby zaczynają pojawiać się w dzieciństwie i nastolatkach i stopniowo rozwijają się przez całe życie, chociaż zespół może dać się odczuć później. Skojarzenie objawów jest zmienne i nie można przewidzieć szybkości postępu choroby ani jej ciężkości.
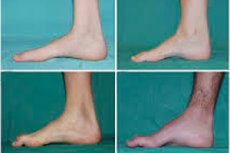
Istnieją takie typowe objawy początkowego etapu, jak zwiększone ogólne zmęczenie; zmniejszone napięcie (osłabienie) mięśni stóp, kostek i podudzi; brak refleksów. Utrudnia to poruszanie stopą i prowadzi do dysbazji (zaburzenia chodu) w postaci wyższego uniesienia nóg, często z częstym potykaniem się i upadkami. Objawami choroby Charcota-Mariego-Tootha u małego dziecka mogą być wyraźne niezdarności i trudności w chodzeniu, niezwykłe dla wieku, związane z obustronnie zwisającą stopą . Charakterystyczne są również deformacje stopy: wysoki łuk (wydrążona stopa) lub mocne płaskie stopy, zakrzywione (młotkowate) palce.
W przypadku chodzenia na palcach u stóp na tle niedociśnienia mięśniowego neurolog może podejrzewać, że dziecko ma CMT typu 4, w której dzieci w wieku młodzieńczym mogą nie być w stanie chodzić.
W miarę postępu atrofii i osłabienia mięśni rozprzestrzeniają się na kończyny górne, utrudniając precyzyjną motorykę i normalną aktywność rąk. Zmniejszenie wrażeń dotykowych i zdolności odczuwania ciepła i zimna, a także drętwienia stóp i dłoni wskazuje na uszkodzenie aksonów nerwów czuciowych.
Z objawami choroby Charcota-Mariego-Tootha w wieku dziecięcym typu 3 i 6 występuje wrażliwa ataksja (upośledzona koordynacja ruchów i równowaga), drżenie i drżenie mięśni, uszkodzenie nerwu twarzowego, zanik nerwu wzrokowego z oczopląsem, utrata słuchu.
Na późniejszych etapach mogą wystąpić niekontrolowane drżenie (drżenie) i częste skurcze mięśni; problemy z ruchem mogą prowadzić do rozwoju bólów: mięśniowych, stawowych, neuropatycznych.
Komplikacje i konsekwencje
Choroba Charcota-Mariego-Tootha może mieć powikłania i konsekwencje, takie jak:
- częstsze skręcenia i złamania;
- przykurcze związane ze skróceniem mięśni i ścięgien okołostawowych;
- skolioza (skrzywienie kręgosłupa);
- problemy z oddychaniem - z uszkodzeniem włókien nerwowych, które unerwiają mięśnie przepony:
- utrata zdolności do samodzielnego poruszania się.
Diagnostyka choroba Charcota-Mariego-Tootha
Diagnostyka obejmuje badanie kliniczne, wywiad (w tym wywiad rodzinny), badanie neurologiczne i systemowe.
Testy są wykonywane w celu sprawdzenia zakresu ruchu, wrażliwości i odruchów ścięgnistych. Przewodnictwo nerwowe można ocenić za pomocą diagnostyki instrumentalnej - elektromiografii lub elektrroneuromyografii . Może być również wymagane USG lub MRI. [6]
Diagnostyka genetyczna lub DNA w celu zidentyfikowania najczęstszych mutacji genetycznych powodujących CMT w próbce krwi jest ograniczona, ponieważ testy DNA nie są obecnie dostępne dla wszystkich typów CMT. Po szczegóły patrz - Badania genetyczne
W niektórych przypadkach wykonuje się biopsję nerwu obwodowego (zwykle mięśnia brzuchatego łydki).
Diagnostyka różnicowa
Diagnozę różnicową przeprowadza się z innymi neuropatiami obwodowymi, dystrofią mięśniową Duchenne'a, zespołami mielopatycznymi i miastenicznymi, neuropatią cukrzycową, z mieloaptiami w przypadku stwardnienia rozsianego i zanikowego bocznego, zespołem Guillaina-Barrégo, urazem nerwu strzałkowego i zanikiem krążka międzykręgowego (w tym kręgosłupa) ), uszkodzenie móżdżku lub wzgórza, a także skutki uboczne chemioterapii (w przypadku leczenia cytostatykami, takimi jak winkrystyna lub paklitaksel). [7]
Z kim się skontaktować?
Leczenie choroba Charcota-Mariego-Tootha
Dziś leczenie tej dziedzicznej choroby polega na ćwiczeniach fizjoterapeutycznych (mających na celu wzmocnienie i rozciągnięcie mięśni); terapia zajęciowa (która pomaga pacjentom z osłabieniem mięśni rąk); używanie urządzeń ortopedycznych ułatwiających chodzenie. Jeśli to konieczne, weź leki przeciwbólowe lub przeciwdrgawkowe. [8]
W przypadku wyraźnych płaskich stóp można wykonać osteotomię, aw przypadku deformacji pięt wskazana jest ich chirurgiczna korekta - artrodeza. [9]
Prowadzone są badania zarówno nad składnikiem genetycznym choroby, jak i metodami jej leczenia. Stosowanie komórek macierzystych, niektórych hormonów, lecytyny czy kwasu askorbinowego nie przyniosło jeszcze pozytywnych rezultatów.
Ale dzięki najnowszym badaniom w niedalekiej przyszłości może naprawdę pojawić się nowe badanie w leczeniu choroby Charcota-Mariego-Tootha. Tak więc od 2014 roku francuska firma Pharnext rozwija, a od połowy 2019 roku badania kliniczne leku PXT3003 do leczenia CMT typu 1 u dorosłych, hamującego zwiększoną ekspresję genu PMP22, poprawiającego mielinizację nerwów obwodowych i osłabienie objawów nerwowo-mięśniowych.
Specjaliści firmy medycznej Sarepta Therapeutics (USA) pracują nad terapią genową choroby Charcota-Marie-Tootha typu 1. Ta terapia będzie wykorzystywać nieszkodliwy wirus związany z adenowirusem (AAV) z rodzaju Dependovirus z liniowym genomem jednoniciowego DNA, który będzie przenosił gen NTF3 do organizmu, który koduje białko neurotrofiny-3 (NT-3) niezbędne do funkcjonowanie komórek nerwowych Schwanna.
Do końca 2020 roku Helixmith rozpocznie badania kliniczne terapii genowej Engensis (VM202) opracowanej w Korei Południowej w celu leczenia objawów mięśniowych w CMT typu 1. [10]
Zapobieganie
Zapobieganie CMT może być poradnictwem genetycznym dla przyszłych rodziców, zwłaszcza jeśli ktoś z małżeństwa ma tę chorobę w rodzinie. Zidentyfikowano jednak przypadki mutacji punktowych genu de novo, czyli przy braku choroby w historii rodziny.
W czasie ciąży pobranie próbki kosmówki kosmówki (od 10 do 13 tygodnia ciąży), a także analiza płynu owodniowego (w wieku 15–18 tygodni) pozwala sprawdzić prawdopodobieństwo wystąpienia choroby Charcota-Mariego-Tootha u nienarodzonego dziecka.
Prognoza
Ogólnie rzecz biorąc, rokowanie dla różnych typów choroby Charcota-Mariego-Tootha zależy od ciężkości klinicznej, ale w każdym przypadku choroba postępuje powoli. Wielu pacjentów jest niepełnosprawnych, chociaż nie zmniejsza to oczekiwanej długości życia.