
Achondroplazja
Ostatnia recenzja: 23.04.2024

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
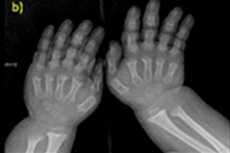
Istnieje wiele rzadkich chorób wrodzonych, a jedną z nich jest naruszenie wzrostu kości - achondroplazja, która prowadzi do wyraźnego nieproporcjonalnego niskiego wzrostu.
W części dotyczącej anomalii rozwojowych ICD-10 kodem tego typu dziedzicznej dysplazji kostno-chrzęstnej z wadami wzrostu kości rurkowych i kręgosłupa jest Q77.4 [1]
Epidemiologia
Statystyki różnych badań dotyczących rozpowszechnienia achondroplazji są niejednoznaczne. Niektórzy twierdzą, że ta anomalia występuje u jednego noworodka na 10 tys., Inni - w jednym z 26-28 tys., A jeszcze inni - 4-15 przypadków na 100 tys. [2]
Istnieją również informacje, że gdy ojciec ma ponad 50 lat, zachorowalność na achondroplazję u dzieci to jeden przypadek na 1875 noworodków.
Przyczyny achondroplazja
Przyczyną achondroplazji jest naruszenie osteogenezy, w szczególności jednego z rodzajów kostnienia wewnątrzmacicznego trzonu kości rurkowych szkieletu - kostnienia wewnątrzchrzęstnego (wewnątrzchrzęstnego), podczas którego następuje modyfikacja chrząstki w tkankę kostną. Po szczegóły patrz - Rozwój i wzrost kości
Naruszenie kostnienia kości długich, czyli achondroplazja płodu następuje z powodu mutacji w genie błonowych kinaz tyrozynowych - receptora 3 czynnika wzrostu fibroblastów (FGFR3 na chromosomie 4p16.3), co wpływa na wzrost i różnicowanie komórek. Obecność mutacji FGFR3 jest związana z niestabilnością genetyczną i zmianami liczby chromosomów (aneuploidia).
Dziecko otrzymuje achondroplazję jako cechę autosomalną dominującą, to znaczy otrzymuje jedną kopię zmutowanego genu (który jest dominujący) i jeden normalny gen na parze chromosomów niepłciowych (autosomalnych). Zatem rodzaj dziedziczenia tej wady jest autosomalny dominujący, a anomalia może objawiać się u 50% potomstwa, gdy krzyżuje się kombinację alleli danego genu (genotypu).
Ponadto mutacje mogą być sporadyczne i, jak pokazuje praktyka, w 80% przypadków dzieci z achondroplazją rodzą się z rodzicami o normalnym wzroście.
Czynniki ryzyka
Główne czynniki ryzyka narodzin dzieci z achondroplazją są dziedziczne. Jeśli jedno z rodziców ma tę wadę, to prawdopodobieństwo urodzenia chorego dziecka szacuje się na 50%; w obecności tej anomalii u obojga rodziców - również 50%, ale z 25% ryzykiem homozygotycznej achondroplazji, prowadzącej do śmierci przed urodzeniem lub we wczesnym dzieciństwie.
Wraz z wiekiem ojca (bliżej 40 lat i więcej) wzrasta ryzyko nowej mutacji (mutacji de novo) genu FGFR3.
Patogeneza
Wyjaśniając patogenezę achondroplazji, eksperci podkreślają znaczenie transbłonowej białkowej kinazy tyrozynowej (kodowanej przez gen FGFR3) w regulacji podziału, różnicowania i apoptozy komórek tkanki chrzęstnej płytek wzrostowych - chondrocytów, a także rozwój szkieletu - osteogeneza i mineralizacja kości.
Podczas rozwoju embrionalnego, przy obecności mutacji genowej, receptory czynnika wzrostu fibroblastów 3 stają się bardziej aktywne. Wzmocnienie ich funkcji zaburza przekazywanie sygnałów komórkowych oraz interakcję pozakomórkowej części tego białka z polipeptydowymi czynnikami wzrostu fibroblastów (FGF). W rezultacie dochodzi do niepowodzenia: etap proliferacji komórek tkanki chrzęstnej staje się krótszy, a ich różnicowanie rozpoczyna się wcześniej niż w przepisanym czasie. Wszystko to prowadzi do niewłaściwego tworzenia się i zespolenia kości czaszki i dysplazji szkieletowej - zmniejszenia kości długich, któremu towarzyszy wyraźny niski wzrost lub karłowatość.
A dwie trzecie przypadków karłowatości wiąże się właśnie z achondroplazją.
Objawy achondroplazja
Nieprawidłowy wzrost kości powoduje takie kliniczne objawy achondroplazji, jak:
- wyraźny niski wzrost (nieproporcjonalny karłowatość) ze średnim wzrostem osoby dorosłej 123-134 cm;
- skrócenie bliższych kończyn dolnych i górnych przy stosunkowo normalnej wielkości tułowia;
- skrócone palce u rąk i nóg;
- powiększona głowa (makro lub megalocefalia); [3]
- specyficzne rysy twarzy w postaci wypukłego czoła i niedorozwoju środkowej części twarzy - obniżony grzbiet nosa.
- wąskie połączenie czaszkowo-szyjne. Niektóre dzieci z achondroplazją umierają w pierwszym roku życia z powodu powikłań związanych z połączeniem czaszkowo-szyjnym; Badania populacyjne pokazują, że to nadmierne ryzyko zgonu może sięgać nawet 7,5% bez oceny i interwencji. [4]
- Dysfunkcja ucha środkowego jest często problemem [5]i, jeśli nie jest odpowiednio leczona, może skutkować przewodzeniową utratą słuchu na tyle ciężką, aby zakłócać rozwój mowy. Ponad połowa dzieci będzie potrzebować rurki wyrównującej ciśnienie. [6] Ogólnie rzecz biorąc, około 40% osób z achondroplazją ma funkcjonalnie znaczną utratę słuchu. Rozwój języka ekspresyjnego jest również często opóźniony, chociaż siła związku między utratą słuchu a problemami z ekspresyjną mową jest wątpliwa.
- skrzywienie nóg jest bardzo częste u pacjentów z achondroplazją. Ponad 90% nieleczonych dorosłych ma pewien stopień skłonności. [7] Uwielbienie jest w rzeczywistości złożoną deformacją, która wynika z połączenia bocznego przechylenia, wewnętrznego skrętu piszczeli i dynamicznej niestabilności kolana. [8]
Niemowlęta z achondroplazją charakteryzują się hipotonią mięśni, dzięki czemu później zaczynają uczyć się ruchu i chodzenia. Ta niepełnosprawność rozwojowa nie wpływa na inteligencję i zdolności poznawcze. [9], [10]
Konsekwencje i komplikacje
W przypadku tego typu dziedzicznej dysplazji kostno-chrzęstnej charakterystyczne są następujące powikłania i konsekwencje:
- nawracające infekcje ucha;
- obturacyjny bezdech senny;
- wodogłowie;
- wady zgryzu i skręcone zęby:
- deformacja nóg (szpotawość lub koślawość) ze zmianą chodu;
- przerostowa lordoza kręgosłupa lędźwiowego lub jego skrzywienie (kifoza piersiowo-lędźwiowa lub skolioza lędźwiowa) - z bólem pleców podczas chodzenia;
- ból stawów (z powodu niewłaściwego umieszczenia kości lub ucisku korzeni nerwowych);
- zwężenie kręgosłupa i ucisk rdzenia kręgowego; Najczęstszą dolegliwością medyczną w wieku dorosłym jest objawowe zwężenie kręgosłupa obejmujące L1-L4. Objawy wahają się od sporadycznego, odwracalnego chromania się wywołanego wysiłkiem fizycznym po ciężkie, nieodwracalne upośledzenie funkcji nóg i trzymanie moczu. [11] Kulawizna i zwężenie mogą powodować zarówno objawy czuciowe (drętwienie, ból, uczucie ciężkości), jak i ruchowe (osłabienie, potykanie się, ograniczona wytrzymałość podczas chodzenia). Kulawizna naczyniowa wynika z obrzęku naczyń krwionośnych po staniu i chodzeniu i ustępuje całkowicie podczas odpoczynku. Zwężenie kręgosłupa jest faktycznym uszkodzeniem rdzenia kręgowego lub korzenia nerwu przez zwężenie kości kanału kręgowego, a objawy są nieodwracalne. Objawy zlokalizowane w określonym dermatomie mogą wynikać ze zwężenia określonych otworów korzeni nerwowych.
- redukcja odcinka piersiowego z ograniczeniem wzrostu płuc i zmniejszeniem ich funkcji (w postaci ciężkiej duszności). W okresie niemowlęcym niewielka grupa osób z achondroplazją ma restrykcyjne problemy z płucami. Małe piersi i zwiększona podatność klatki piersiowej łącznie powodują zmniejszenie objętości płuc i restrykcyjną chorobę płuc [12]
Inne problemy ortopedyczne
- Słabość stawów. Większość stawów jest hipermobilna w dzieciństwie. Ogólnie ma to niewielki wpływ, z wyjątkiem niestabilności kolana u niektórych osób.
- Łąkotka boczna tarczowa. Ta nowo zidentyfikowana nieprawidłowość strukturalna może u niektórych osób prowadzić do przewlekłego bólu kolana. [13]
- Artretyzm. Konstytutywna aktywacja FGFR-3, podobnie jak w przypadku achondroplazji, może chronić przed rozwojem zapalenia stawów. [14]
- Acanthosis nigricans występuje u około 10% osób z achondroplazją. [15]W tej populacji nie odzwierciedla to hiperinsulinemii ani nowotworu złośliwego.
Achondroplazja homozygotyczna, wywoływana przez bialleliczne, patogenne warianty nukleotydu 1138 FGFR3, jest poważną chorobą ze zmianami radiologicznymi, które różnią się jakościowo od achondroplazji. Wczesna śmierć następuje w wyniku niewydolności oddechowej z powodu małej klatki piersiowej oraz ubytków neurologicznych w wyniku zwężenia szyjno-rdzeniowego [Hall 1988].
Diagnostyka achondroplazja
U większości pacjentów achondroplazję rozpoznaje się na podstawie charakterystycznych objawów klinicznych i wyników radiologicznych. U niemowląt lub w przypadku braku pewnych objawów do dokładnej diagnozy stosuje się badanie genetyczne - badanie kariotypu . [16]
Przeprowadzając diagnostykę prenatalną z wykorzystaniem genetyki molekularnej, można wykonać badania na próbce płynu owodniowego (owodniowego) lub kosmówki kosmówkowej.
Oznaki achondroplazji w USG płodu - skrócenie kończyn i typowe rysy twarzy - są uwidaczniane po 22. Tygodniu ciąży.
Diagnostyka instrumentalna obejmuje również RTG szkieletu czy USG kości . Radiografia potwierdza diagnozę na podstawie danych, takich jak duża czaszka z wąskim otworem potylicznym i stosunkowo małą podstawą; krótkie rurkowate kości i skrócone żebra; krótkie i spłaszczone kręgosłupy; zwężony kanał kręgowy, zmniejszony rozmiar skrzydeł biodrowych.
Diagnostyka różnicowa
Konieczna jest diagnostyka różnicowa z karłowatością przysadkową (karłowatością) , wrodzoną dysplazją spondyloepiphyseal i diastroficzną, hipochondroplazją, zespołami Shereshevsky-Turnera i Noonana, pseudoachondroplazją. Tak więc różnica między pseudoachondroplazją a achondroplazją polega na tym, że u pacjentów z karłowatością z pseudoachondroplazją wielkość głowy i rysy twarzy są normalne.
Z kim się skontaktować?
Leczenie achondroplazja
Zalecenia dotyczące monitorowania zdrowia dzieci z achondroplazją zostały określone przez Komitet Genetyki Amerykańskiej Akademii Pediatrycznej. Te zalecenia służą jako wskazówki i nie zastępują podejmowania indywidualnych decyzji. Niedawna recenzja [Pauli & Botto 2020] zawiera również wskazówki od przewodnika. Istnieją wyspecjalizowane kliniki zajmujące się leczeniem dysplazji szkieletowej; ich zalecenia mogą się nieznacznie różnić od tych ogólnych zaleceń.
Zalecenia obejmują (ale nie są do nich ograniczone) następujące elementy.
Wodogłowie. Jeśli pojawią się oznaki lub objawy zwiększonego ciśnienia śródczaszkowego (np. Przyspieszony wzrost głowy, stale wypukłe ciemiączko, zauważalny wzrost wypukłości żył powierzchownych na twarzy, drażliwość, wymioty, zmiany widzenia, ból głowy), skierowanie do lekarza neurochirurg jest konieczny.
Zakładaną etiologią wodogłowia w achondroplazji jest podwyższone śródczaszkowe ciśnienie żylne na skutek zwężenia otworu szyjnego. Dlatego standardowym leczeniem było przecieranie komorowo-otrzewnowe. Jednak endoskopowa trzecia ventriculostomia może być korzystna u niektórych osób, [17]co oznacza, że inne mechanizmy, takie jak blokowanie ujść komór z powodu zwężenia czaszkowo-szyjnego, mogą być przydatne. [18]
Zwężenie połączenia czaszkowo-szyjnego. Najlepszymi predyktorami potrzeby dekompresji podpotylicznej są:
- Hiperrefleksja lub klonus kończyn dolnych
- Centralny spłycenie oddechu w polisomnografii
- Zmniejszenie wielkości otworu wielkiego, określone przez tomografię komputerową połączenia czaszkowo-szyjnego i porównane z normami dla dzieci z achondroplazją. [19]
- Oznaki ucisku rdzenia kręgowego i / lub nieprawidłowości sygnału ważonego T 2; ostatnio zasugerowano jako kolejny czynnik, który należy wziąć pod uwagę przy podejmowaniu decyzji o pracy.
Jeśli są wyraźne oznaki objawowego ucisku, należy pilnie skontaktować się z neurochirurgiem dziecięcym w celu przeprowadzenia operacji dekompresyjnej. [20]
Obturacyjny bezdech senny. Leczenie może obejmować:
- Adenotonsillektomia
- Dodatnie ciśnienie w drogach oddechowych
- Tracheostomia w skrajnych przypadkach
- Zmniejszenie wagi
Te interwencje mogą prowadzić do poprawy zaburzeń snu i pewnej poprawy funkcji neurologicznych. [21]
W rzadkich przypadkach, gdy niedrożność jest na tyle poważna, że uzasadnia tracheostomię, w celu złagodzenia niedrożności górnych dróg oddechowych zastosowano operację przesunięcia środkowej części twarzy. [22]
Dysfunkcja ucha środkowego. W razie potrzeby należy agresywnie leczyć częste infekcje ucha środkowego, uporczywy płyn w uchu środkowym i późniejszą utratę słuchu. Rurki długo działające są zalecane, ponieważ często są potrzebne do siódmego lub ósmego roku życia. [23]
Jeśli w jakimkolwiek wieku pojawią się problemy, zaleca się zastosowanie odpowiednich metod leczenia.
Niski wzrost. W wielu badaniach oceniano terapię hormonem wzrostu (GH) jako możliwą metodę leczenia krótkiej achondroplazji. [24]
Ogólnie te i inne serie wykazują początkowe przyspieszenie wzrostu, ale jego efekt słabnie z czasem.
Średnio można spodziewać się wzrostu wzrostu osoby dorosłej tylko o około 3 cm.
Wydłużone wydłużanie kończyny różnymi technikami pozostaje opcją dla niektórych. Możesz zwiększyć swój wzrost nawet o 30-35 cm [25] Powikłania są częste i mogą być poważne.
Chociaż niektórzy zwolennicy wykonywania tych procedur już w wieku od sześciu do ośmiu lat, wielu pediatrów, genetyków klinicznych i etyków opowiada się za odroczeniem takiej operacji do czasu, gdy młoda osoba będzie mogła uczestniczyć w podjęciu świadomej decyzji.
Przynajmniej w Ameryce Północnej tylko niewielka część osób dotkniętych chorobą decyduje się na wydłużenie kończyn. Medyczna Rada Doradcza Little People of America wydała oświadczenie dotyczące stosowania wydłużonego wydłużania kończyn.
Otyłość. Środki zapobiegające otyłości należy rozpocząć we wczesnym dzieciństwie. Standardowe metody leczenia otyłości powinny być skuteczne u osób z achondroplazją, chociaż zapotrzebowanie kaloryczne jest mniejsze. [26]
Do śledzenia postępów należy stosować standardowe siatki masy ciała i masy wzrostu specyficzne dla achondroplazji. Należy zauważyć, że te krzywe nie są idealnymi krzywymi wagi do wzrostu; uzyskano je z tysięcy punktów danych od osób z achondroplazją.
Standardy wskaźnika masy ciała (BMI) zostały opracowane dla dzieci w wieku do 16 lat. [27]BMI nie jest standaryzowane dla dorosłych z achondroplazją; porównanie z krzywymi BMI dla średniego wzrostu da błędne wyniki. [28]
Deformacja szpotawości. Coroczna kontrola ortopedyczna jest zalecana przez lekarza zaznajomionego z achondroplazją lub chirurga ortopedy. Opublikowano kryteria chirurgiczne. [29]
Obecność postępującego objawowego zgięcia wymaga skierowania do podiatry. Sama deformacja szpotawości bez objawów zwykle nie wymaga korekcji chirurgicznej. Można wybrać różne interwencje (np. Sterowany wzrost przy użyciu ośmiu płytek, osteotomia koślawości i osteotomia derotacyjna). Nie ma kontrolowanych badań porównujących wyniki opcji leczenia.
Kifoza. Niemowlęta z achondroplazją często rozwijają elastyczną kifozę. Dostępny jest protokół zapobiegający rozwojowi stałej kifozy kątowej, który obejmuje unikanie wózków z elastycznym oparciem, huśtawek i noszenia. Rada przeciwko siedzeniu bez wsparcia; trzymaj dziecko zawsze za plecami.
- Kifoza poprawia się lub ustępuje u większości dzieci po ortogrodzie i chodzeniu. [30]
- U dzieci, które nie mają samoistnej remisji po zwiększeniu siły tułowia i rozpoczęciu chodzenia, fiksacja jest zwykle wystarczająca, aby zapobiec utrzymywaniu się kifozy piersiowo-lędźwiowej. [31]
- Jeśli ciężka kifoza utrzymuje się, może być konieczna operacja kręgosłupa, aby zapobiec powikłaniom neurologicznym. [32]
Zwężenie kręgosłupa. Jeśli pojawią się poważne oznaki i / lub objawy zwężenia kręgosłupa, konieczne jest pilne skierowanie do specjalisty chirurga.
Zwykle zalecana jest szeroka i szeroka laminektomia. Trafność zabiegu zależy od poziomu (np. Klatki piersiowej lub odcinka lędźwiowego) i stopnia zwężenia. Pacjenci mieli lepsze wyniki i lepszą funkcję niż wcześniej przeszli operację po wystąpieniu objawów [33]
Immunizacja. Nic na temat achondroplazji nie wyklucza wszystkich rutynowych szczepień. Ze względu na zwiększone ryzyko oddechowe szczególnie ważne są szczepionki DTaP, przeciw pneumokokom i grypie.
Potrzeby adaptacyjne. Ze względu na niewielki wzrost konieczne są zmiany środowiskowe. W szkole może to obejmować stołki, obniżone włączniki światła, toalety o odpowiedniej wysokości lub inne środki dostępu, niższe ławki i podnóżki przed krzesłami. W nagłych przypadkach wszystkie dzieci powinny mieć możliwość samodzielnego opuszczenia budynku. Małe dłonie i słabe więzadła mogą utrudniać zdolności motoryczne. Odpowiednie adaptacje obejmują użycie mniejszych klawiatur, cięższych stalówek i gładszych powierzchni do pisania. Większość dzieci musi mieć IEP lub 504.
Do jazdy prawie zawsze potrzebne są przedłużenia pedałów. Mogą być również wymagane modyfikacje przestrzeni roboczej, takie jak niższe biurka, mniejsze klawiatury, stopnie i dostęp do toalety.
Socjalizacja. Ze względu na bardzo zauważalny niski wzrost związany z achondroplazją, chorzy i ich rodziny mogą mieć trudności z socjalizacją i adaptacją do szkoły.
Grupy wsparcia, takie jak Little People of America, Inc. (LPA), mogą pomóc rodzinom rozwiązać te problemy poprzez wsparcie rówieśnicze, przykład i programy świadomości społecznej.
Informacje o zatrudnieniu, edukacji, prawach osób niepełnosprawnych, adopcji małych dzieci, problemach zdrowotnych, odpowiedniej odzieży, adaptacyjnym przystosowaniu i rodzicielstwie są dostępne za pośrednictwem krajowego biuletynu, seminariów i warsztatów.
Nie ma leków, a także środków nieleczniczych, które mogłyby wyleczyć tę wrodzoną wadę rozwojową.
Najczęściej stosowany zabieg fizjoterapeutyczny; może również wymagać leczenia wodogłowia (wszczepienie bajpasu lub endoskopowa ventriculostomia), otyłości, [34]bezdechu sennego, [35]infekcji ucha środkowego lub zwężenia kręgosłupa.
W niektórych klinikach po ukończeniu przez dziecko od pięciu do siedmiu lat podejmuje się leczenie operacyjne: wydłużenie kości podudzia, ud, a nawet kości ramiennej lub korekcję deformacji - operacjami i specjalnymi urządzeniami ortopedycznymi - w trzech do czterech etapach, z czas trwania każdego do 6-12 miesięcy...
Terapia fazy badawczej
Wprowadzenie analogu peptydu natriuretycznego typu C jest w trakcie badań klinicznych. Wczesne wyniki wykazały, że był on dobrze tolerowany i powodował przyspieszenie wzrostu w stosunku do wartości wyjściowej u dzieci z achondroplazją ( ośrodek badawczy ). [36]Skoniugowany peptyd natriuretyczny typu C jest również obecnie poddawany próbom klinicznym ( сайт испытанийośrodek badawczy ). [37]Inne rozważania obejmują hamowanie kinazy tyrozynowej [38], meklizynę [39]i rozpuszczalny rekombinowany ludzki wabik FGFR3. [40]
Przeszukaj Clinicaltrials.gov w amerykańskim i europejskim rejestrze badań klinicznych w Europie, aby uzyskać informacje na temat badań klinicznych dotyczących szerokiego zakresu chorób i stanów.
Zapobieganie
Jedynym środkiem zapobiegawczym jest diagnostyka prenatalna chorób wrodzonych . [41], [42]
Prognoza
Jak długo żyją ludzie z achondroplazją? Średnia długość życia jest o 10 lat krótsza.
Ponieważ zmiany patologiczne w tkance kostnej i stawach prowadzą do ograniczenia samoopieki i ruchu, dzieci z tą diagnozą otrzymują status osób niepełnosprawnych. W dłuższej perspektywie rokowanie u większości pacjentów jest prawidłowe, ale wraz z wiekiem istnieje zwiększone ryzyko chorób serca. [43]