Ekspert medyczny artykułu

Nowe publikacje
Niedokrwistość przysadkowa (karłowatość)
Ostatnia recenzja: 12.07.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Termin „karłowatość przysadkowa” (od greckiego nanos – karzeł; syn.: karłowatość, nanosomia, mikrosomia) w sensie bezwzględnym oznacza chorobę, której głównym objawem jest gwałtowne zahamowanie wzrostu, związane z zaburzeniem wydzielania hormonu wzrostu przez przedni płat przysadki mózgowej.
W szerszym ujęciu karłowatość jest zaburzeniem wzrostu i rozwoju fizycznego, którego wystąpienie może być spowodowane nie tylko bezwzględnym lub względnym niedoborem hormonu somatotropowego spowodowanym patologią samej przysadki mózgowej, ale także zaburzeniem podwzgórzowej (mózgowej) regulacji jej funkcji, defektami w biosyntezie hormonu somatotropowego i zaburzeniami wrażliwości tkanek na ten hormon.
 [
[ Przyczyny karłowatość
Większość form karłowatości przysadkowej to choroby genetyczne. Najczęstszą jest karłowatość panhipopituitarna, która dziedziczona jest głównie w sposób recesywny. Przyjmuje się, że istnieją 2 typy transmisji tej formy patologii - autosomalna i poprzez chromosom X. W tej formie karłowatości, wraz z defektem wydzielania hormonu somatotropowego, najczęściej zaburzone jest wydzielanie gonadotropin i hormonu tyreotropowego. Wydzielanie ACTH jest zaburzone rzadziej i w mniejszym stopniu. Badania funkcjonalne z hormonami uwalniającymi, w tym syntetycznym hormonem uwalniającym somatotropinę (składającym się z 29, 40 i 44 reszt aminokwasowych), podobnym do polipeptydu trzustkowego, wykazały, że większość tych pacjentów ma patologię na poziomie podwzgórza, a niewydolność przedniego płata przysadki mózgowej jest wtórna. Pierwotna patologia samej przysadki mózgowej jest mniej powszechna.
Genetyczna karłowatość z izolowanym niedoborem hormonu wzrostu, z upośledzoną aktywnością biologiczną i wrażliwością na niego, spotykana jest sporadycznie w Rosji i krajach sąsiednich. Jest bardziej powszechna na kontynencie amerykańskim, w krajach Bliskiego i Środkowego Wschodu oraz w Afryce. Na podstawie wyników badania zawartości hormonu somatotropowego we krwi i wrażliwości pacjentów na egzogenny hormon somatotropowy, poziomu immunoreaktywnej insuliny (IRI), insulinopodobnych czynników wzrostu (IGF) typu I (somatomedyny C) i typu II oraz odpowiedzi IGF-1 na leczenie preparatami hormonu somatotropowego, zidentyfikowano różne warianty klinicznie podobnych typów karłowatości.
Niedawno wyjaśniono patogenezę karłowatości Larona, która jest spowodowana niedoborem IRF-1 i IRF-II, a także patogenezę karłowatości u afrykańskich pigmejów, związaną z niedoborem IRF-II.
W 1984 roku opisano nową odmianę karłowatości pseudoprzysadkowej z wysokim poziomem hormonu somatotropowego i IGF-1; genezę karłowatości tłumaczy się defektem jej receptorów, o czym świadczy gwałtowny spadek wiązania fibroblastów skóry z IGF-1.
Należy podkreślić, że we współczesnych warunkach, przy występowaniu małych rodzin, wiele odosobnionych („idiopatycznych”, sporadycznych) przypadków choroby może mieć również podłoże genetyczne.
W analizie 350 historii przypadków etiologia karłowatości była niejasna u 228 pacjentów (65,2%). Do tej grupy należeli pacjenci z 57 rodzin z powtarzającym się występowaniem karłowatości (2-4 przypadki na rodzinę), co stanowiło 28% wszystkich pacjentów. W 77% rodzin z niejasnymi etiologicznie (głównie genetycznymi) formami karłowatości ustalono niezaprzeczalny związek z dziedziczeniem braku czynnika Rh. Rozkład czynnika Rh w rodzinach pacjentów z karłowatością różni się od obserwowanego w konflikcie Rh między matką a płodem i z reguły nie towarzyszy mu choroba hemolityczna noworodka (ojciec może być Rh ujemny, a w przypadku heterozygotyczności rodziców dla czynnika Rh - dzieci itp.). Można założyć związek między aktywnością genów odpowiedzialnych za syntezę hormonu somatotropowego (lub hormonu uwalniającego somatotropinę) a genami determinującymi czynnik Rh, zwłaszcza że większość form karłowatości i brak czynnika Rh to cechy autosomalne recesywne. Nie wyjaśnia to względnej rzadkości karłowatości w porównaniu z częstością występowania osób Rh-ujemnych w populacji. Prawdopodobnie ważne są pewne, jak dotąd nieznane, dodatkowe czynniki, ale cechy rozkładu czynnika Rh w rodzinach pacjentów z karłowatością rodzinną i sporadyczną raczej nie są przypadkowe.
Dużą grupę pacjentów z karłowatością (pierwotną mózgową, mózgowo-przysadkową) stanowią pacjenci z różnymi typami patologii organicznych ośrodkowego układu nerwowego, które powstały w życiu płodowym lub we wczesnym dzieciństwie. Podłożem anatomicznym powodującym tę patologię może być niedorozwój lub brak przysadki mózgowej, jej dystopia w patologii powstawania siodła tureckiego, zwyrodnienie torbielowate przysadki mózgowej, jej zanik spowodowany uciskiem przez guz (czaszkogardlak, gruczolak chromofobowy, oponiak, glejak). Karłowatość może być spowodowana urazami okolicy podwzgórzowo-przysadkowej (wewnątrzmacicznym, porodowym lub poporodowym), do których często dochodzi w ciążach mnogich, a także podczas porodu miednicowego, w ułożeniu stóp lub w położeniu poprzecznym z rotacją na nodze (jest to mechanizm porodu u ponad 1/3 pacjentów z karłowatością). Istotne są uszkodzenia zakaźne i toksyczne (wewnątrzmaciczne zakażenia wirusowe, gruźlica, kiła, malaria, toksoplazmoza; choroby w młodym wieku, posocznica noworodkowa, zapalenie opon mózgowych i pajęczaków itp.). Procesy te mogą uszkodzić samą przysadkę mózgową, ośrodki podwzgórza, które regulują jej funkcję, i zakłócić normalne połączenia funkcjonalne w ośrodkowym układzie nerwowym.
Wewnątrzmaciczne uszkodzenia płodu mogą doprowadzić do urodzenia pacjentek z „karłowatością od urodzenia” z prawidłowym wydzielaniem hormonu wzrostu (karłowatość pierwotna mózgowa, małogłowie, karłowatość Silvera-Russella z hemiasymetrią ciała i wysokim poziomem gonadotropin itp.).
Dodatkowymi czynnikami pogłębiającymi zaburzenia rozwoju fizycznego w karłowatości mogą być niedostateczne odżywianie, niezrównoważone pod względem niezbędnych składników (niedobór białka) i mikroelementów (niedobór cynku) oraz niekorzystne warunki środowiskowe, a także różne choroby przewlekłe, takie jak kłębuszkowe zapalenie nerek, w którym azotemia wpływa na aktywność receptorów wątrobowych lub bezpośrednio wpływa na metabolizm komórek wątroby, powodując spadek syntezy somatomedyny, lub marskość wątroby, gdy powstawanie somatomedyny jest upośledzone.
Patogeneza
U większości chorych z karłowatością przysadkową zmiany nie ograniczają się do patologii wydzielania hormonu somatotropowego i wrażliwości na niego, ale obejmują również inne hormony tropowe przysadki, co powoduje różnorodne kombinacje zaburzeń endokrynologicznych i metabolicznych.
W izolowanym niedoborze hormonu somatotropowego zmiany morfologiczne w przysadce mózgowej są słabo zbadane. W badanych przypadkach rzadko stwierdzano zaburzenia patologiczne (kraniopharyngioma lub hiperostoza kości czaszki). W tym typie karłowatości można zaobserwować wrodzony niedorozwój komórek peptydergicznych lub defekt układu neuroprzekaźników w podwzgórzu. W takich przypadkach karłowatość może być połączona z dysplazją lub hipoplazją nerwów wzrokowych. Torbiele wewnątrzsiodłowe, guzy przysadki i podwzgórza prowadzą do niedoboru STH, powodując ucisk tkanki przysadki, w szczególności somatotropów.
Karłowatość charakteryzuje się ścieńczeniem kości, głównie z powodu warstwy korowej, opóźnionego różnicowania i kostnienia szkieletu. Narządy wewnętrzne są hipoplastyczne, czasami zanikowe, a mięśnie słabo rozwinięte.
Objawy karłowatość
Głównymi objawami karłowatości przysadkowej są gwałtowne opóźnienie wzrostu i rozwoju fizycznego. Pacjenci rodzą się z prawidłową masą ciała i długością, a opóźnienie wzrostu zaczyna się w wieku 2-4 lat.
Przed wprowadzeniem aktywnego leczenia nanizmu karły były uważane za te, których wzrost był mniejszy niż 120 cm dla kobiet i 130 cm dla mężczyzn. Obecnie wzrost karła różni się o co najmniej 2-3 sigma od średniej normy tabelarycznej dla danej płci, wieku lub populacji. Istnieje również metoda graficznej oceny wzrostu oparta na krzywej rozkładu Gaussa. W tym przypadku karły według wzrostu są uwzględniane w grupie, która obejmuje minimalną liczbę osobników w odpowiedniej populacji z największym opóźnieniem od średniej normy wzrostu.
Karłowatość przysadkowa charakteryzuje się nie tylko małą bezwzględną masą ciała, ale także małą roczną dynamiką wzrostu i rozwoju fizycznego. Budowa ciała jest proporcjonalna, ale proporcje ciała chorych są typowe dla dzieciństwa. Skóra jest blada, często o żółtawym odcieniu, sucha, co jest spowodowane bezwzględną lub względną niedoczynnością tarczycy, czasami obserwuje się sinicę - „marmurkowanie” skóry. U nieleczonych pacjentów wcześnie pojawia się staro wyglądająca i pomarszczona skóra (geroderma). Jest to spowodowane niedoborem anabolicznego działania STH i powolną zmianą generacji komórkowych.
Włosy na głowie mogą być normalne lub suche, cienkie, łamliwe; typowe są długie rzęsy. Często nie występuje wtórny wzrost włosów. Wielkość siodła tureckiego u większości pacjentów z karłowatością (70-75%) nie ulega zmianie, ale siodło często zachowuje dziecięcy kształt „stojącego owalu”, ma szerokie „młodzieńcze” plecy, zatoka kości klinowej pozostaje w tyle pod względem pneumatyzacji. Są jednak pacjenci z powiększonym siodłem tureckim, co jest oznaką guza; z obszarami zwapnienia na jego tle lub w okolicy wejścia (w przypadku kraniopharyngioma, resztkowych skutków neuroinfekcji) lub jego zmniejszeniem (objawy niedorozwoju, małych rozmiarów przysadki mózgowej). Objawy nadciśnienia wewnątrzczaszkowego są obserwowane: ścieńczenie kości sklepienia czaszki, zwiększony wzór naczyniowy, obecność odcisków palców itp. Najważniejszym objawem karłowatości przysadkowej jest opóźnienie w czasie różnicowania i kostnienia szkieletu. Cechy układu zębowego są również ściśle związane z różnicowaniem szkieletowym: obserwuje się późną wymianę zębów mlecznych. Największe opóźnienie w rozwoju układu kostnego obserwuje się u pacjentów z karłowatością z niewydolnością seksualną i niedoczynnością tarczycy.
Narządy płciowe większości pacjentów są poważnie niedorozwinięte, chociaż wady rozwojowe są rzadkie. Zaobserwowaliśmy wnętrostwo u 5,8% pacjentów płci męskiej. Niewydolność seksualna jest połączona z niedorozwojem drugorzędnych cech płciowych i zmniejszonym pożądaniem seksualnym, brakiem miesiączki. Prawidłowy spontaniczny rozwój seksualny jest obserwowany tylko u pacjentów z izolowanym niedoborem hormonu wzrostu i u niektórych pacjentów z karłowatością mózgową.
Niedoczynność tarczycy jest dość powszechnym objawem karłowatości. Należy zauważyć, że zewnętrzne objawy niedoczynności tarczycy nie zawsze odzwierciedlają prawdziwy stan czynnościowy tarczycy. Wynika to ze względnej niedoczynności tarczycy spowodowanej zaburzeniem przejścia tyroksyny (T 4 ) w trójjodotyroninę (T 3 ) i powstawaniem nieaktywnej (odwracalnej) T 3, co jest charakterystyczne dla niedoczynności somatotropowej.
Funkcja adrenokortykotropowa w karłowatości przysadkowej zmniejsza się rzadziej i w mniejszym stopniu niż funkcje stymulujące czynność płciową i tarczycę, a u większości pacjentów nie wymaga specjalnej korekty.
W większości przypadków intelekt nie jest upośledzony. Spotykane są zmiany emocjonalne w postaci infantylizmu umysłowego; u starszych pacjentów bez upośledzenia intelektualnego obserwuje się niekiedy nerwice reaktywne.
W organicznych patologiach mózgu, zwłaszcza o charakterze nowotworowym, karłowatość może występować z objawami moczówki prostej, niedowidzenia hemianopsyjnego dwuskroniowego i niepełnosprawności intelektualnej.
Badanie rozwoju aktywności bioelektrycznej mózgu u pacjentów bez organicznych objawów ośrodkowego układu nerwowego wykazało, że ich EEG charakteryzuje się cechami niedojrzałości, długotrwałym zachowaniem wysokiego „dziecinnego” napięcia EEG; nierównomiernością amplitudy i częstotliwości rytmu alfa; gwałtownym wzrostem zawartości wolnych rytmów θ i δ, szczególnie w odprowadzeniach czołowych i centralnych; wyraźną reakcją na hiperwentylację; przesunięciem zakresu rytmów EEG po rytmach stymulacji świetlnej w stronę niskich częstotliwości (dowód na zmniejszenie ruchomości funkcjonalnej struktur nerwowych mózgu). Ujawniono, że u starszych pacjentów niedojrzały charakter aktywności elektrycznej mózgu wynika z niedorozwoju seksualnego, a u pacjentów we wszystkich grupach wiekowych – z niedoczynności tarczycy.
Metabolizm węglowodanów u pacjentów z karłowatością charakteryzuje się tendencją do obniżania się poziomu glukozy we krwi na czczo, wzrostem podczas wysiłku fizycznego, niedoborem endogennej insuliny, zwiększoną wrażliwością na insulinę egzogenną z częstym rozwojem stanów hipoglikemicznych. Te ostatnie tłumaczy się głównie niedostatecznym poziomem hormonów przeciwwyspowych w organizmie pacjentów.
Narządy wewnętrzne wykazują splanchnomycrię, tj. zmniejszenie ich rozmiarów. Nie opisano żadnych zmian czynnościowych w narządach wewnętrznych specyficznych dla karłowatości. Często obserwuje się niedociśnienie tętnicze ze zmniejszonym ciśnieniem skurczowym i rozkurczowym oraz zmniejszoną amplitudą tętna. Tony serca są stłumione, słyszalne są szmery czynnościowe o różnej tematyce z powodu zmian troficznych w mięśniu sercowym i zaburzeń autonomicznych. EKG charakteryzuje się niskim napięciem (szczególnie w przypadku niedoczynności tarczycy), bradykardią zatokową lub bradyarytmią; PCG wykazuje zmniejszenie amplitudy tonów, tonów dodatkowych i szmerów czynnościowych. Dane oksygemometryczne wskazują na hipoksemię (początkową i podczas wysiłku fizycznego) i dług tlenowy. U pacjentów w podeszłym wieku czasami rozwija się nadciśnienie.
Diagnostyka karłowatość
Diagnozę i różnicowanie karłowatości ustala się na podstawie danych z wywiadu oraz kompleksowego badania klinicznego, radiologicznego, laboratoryjnego i hormonalnego. Oprócz bezwzględnej masy ciała, w celu oceny wzrostu pacjentów, określa się deficyt wzrostu - różnicę między wzrostem pacjenta a jego średnią normą dla odpowiedniej płci i wieku; wiek wzrostu - zgodność wzrostu pacjenta z określonymi normami; wskaźnik odchylenia znormalizowanego
I = M - Mcp / δ, gdzie M to wzrost pacjenta, Mcp to średni wzrost prawidłowy dla danej płci i wieku, δ to odchylenie kwadratowe od Mcp; I mniejsze niż 3 jest typowe dla nanizmu, I większe niż 3 jest typowe dla gigantyzmu. Wskaźnik ten można wykorzystać do oceny dynamiki rozwoju.
Badanie rentgenowskie pacjentów z karłowatością ujawnia objawy nadciśnienia wewnątrzczaszkowego, resztkowe skutki neuroinfekcji, zwapnienia i kraniosynostozę. Badanie wielkości, kształtu i struktury siodła tureckiego jest uważane za pośredni wskaźnik charakteryzujący wielkość przysadki mózgowej. Jednym z najważniejszych objawów patologicznego zahamowania wzrostu jest zaburzenie różnicowania szkieletowego. Aby ocenić stopień dojrzałości szkieletu, określa się wiek kostny (radiograficzny), który odpowiada zróżnicowaniu tkanki kostnej; niedobór kostnienia to stopień opóźnienia kostnienia od normy (w latach), współczynnik kostnienia to iloraz podzielenia wieku kostnego przez parametry chronologiczne i inne.
Współczesna diagnostyka karłowatości jest niemożliwa bez zbadania wydzielania hormonu somatotropowego, jego podstawowego poziomu, rytmu dobowego i uwalniania pod wpływem stymulacji. U większości pacjentów z karłowatością przysadkową obserwuje się obniżoną zawartość hormonu somatotropowego w surowicy krwi. Przy oznaczaniu metodą radioimmunologiczną wynosi ona (według różnych autorów) od (0,87±0,09) do (1,50±0,64) ng/ml, przy średniej normie (3,81±0,29) ng/ml. Badanie dobowych (dobowych) rytmów wydzielania hormonu somatotropowego wykazało, że jego poziom u zdrowych osób jest maksymalny w ciągu pierwszych 2 godzin snu oraz o godzinie 4-6 rano. W karłowatości zawartość hormonu somatotropowego jest obniżona również w tych godzinach.
Do badania rezerw funkcji somatotropowej stosuje się różne środki pobudzające, badając zawartość hormonu somatotropowego przed i po ich podaniu. Krew do badania pobiera się co 30 minut przez 2-3 godziny. Wydzielanie hormonu somatotropowego po stymulacji uważa się za prawidłowe przynajmniej do 7-10 ng/ml, czasami osiąga 20-40 ng/ml. Jeśli w jednej z próbek nie ma reakcji, przeprowadza się powtórne testy z innymi środkami pobudzającymi. Niedobór hormonu somatotropowego uważa się za udowodniony w przypadku braku wydzielania hormonu somatotropowego w 2-3 różnych próbkach.
Najczęściej stosowane testy stymulujące to: przy dożylnym podaniu 0,1 U (0,75-1,5 U) insuliny na 1 kg masy ciała pacjenta i osiągnięciu hipoglikemii (obniżenie poziomu glukozy we krwi o 50% w porównaniu do poziomu wyjściowego), oznacza się poziom hormonu somatotropowego w surowicy według powyższego schematu. W przypadku wystąpienia ciężkiej hipoglikemii badanie przerywa się, a pacjentowi podaje się glukozę dożylnie. Jest to najpowszechniejsza, klasyczna metoda diagnostyczna.
TRH w dawce 200-500 mcg dożylnie. Skutecznie identyfikuje rezerwy hormonalne, nie powoduje powikłań. W połączeniu z testem insulinowym pozwala ocenić stopień uszkodzenia układu podwzgórzowo-przysadkowego. Pozytywna reakcja na TRH przy braku hipoglikemii na insulinę wskazuje na nienaruszoność przysadki i uszkodzenie na poziomie podwzgórza, negatywne reakcje na TRH i hipoglikemia wskazują na uszkodzenie samej przysadki.
TRH, LH-RH w dawce 300 mcg dożylnie działa podobnie do poprzedniego.
Ludzki SGH jest syntetycznym analogiem biologicznie czynnego związku wyizolowanego z guza trzustki. Obecnie istnieją 3 rodzaje syntetycznego SGH: z 29, 40 i 44 resztami aminokwasowymi. Stosuje się go dożylnie w dawkach od 1 do 3 μg/kg masy ciała pacjenta. Uwalnianie STH obserwuje się po 15-20 minutach od podania, test jest skuteczniejszy od innych w ujawnianiu rezerw endogennego hormonu wzrostu. Pozytywna reakcja SGH wskazuje na podwzgórzowy poziom uszkodzenia funkcji somatotropowej i nienaruszoną przysadkę mózgową; z aminokwasami (monochlorek L-argininy, ornityna, tryptofan, glicyna, leucyna) dożylnie w dawce 0,25-0,5 g na 1 kg masy ciała pacjenta. Skuteczny w badaniu rezerw SGH. Może powodować reakcje alergiczne.
L-dopa doustnie w dawce 250-500 mcg. Skuteczna, dobrze tolerowana przez pacjentów.
Stosuje się również testy z glukagonem, bromową ergokryptyną (parlodel), lizynową wazopresyną, klonidyną oraz dawkowane obciążenie ergometryczne na rowerze stacjonarnym.
Badanie stanu funkcji somatotropowej jest konieczne nie tylko dla diagnostyki karłowatości, ale także dla uzasadnionego wyboru metody terapii, gdyż leczenie somatotropiną jest racjonalne jedynie w przypadku niedoboru endogennego hormonu wzrostu.
Do diagnozy postaci karłowatości bardzo ważne jest zbadanie zawartości insulinopodobnych czynników wzrostu, czyli somatomedyn (zwłaszcza IGF-1, czyli somatomedyny C) - mediatorów działania hormonu somatotropowego na poziomie tkankowym. Wiadomo, że zawartość somatomedyny C w karłowatości jest obniżona, a w akromegalii - zwiększona w porównaniu z normą. Postać karłowatości opisana przez Larona jest rodzajem choroby z prawidłową produkcją STH, ale z zaburzeniem powstawania IGF-1 i IGF-II. Leczenie takich pacjentów somatotropiną jest bezcelowe.
Pośrednimi wskaźnikami somatotropowej funkcji przysadki mózgowej są aktywność fosfatazy alkalicznej i zawartość fosforu nieorganicznego w surowicy. W stanach hiposomatotropowych wskaźniki te są obniżone. W panhipopituitarnej postaci karłowatości następuje zmniejszenie wydzielania gonadotropin, często TSH, czemu towarzyszy odpowiednie zmniejszenie funkcji gruczołów płciowych (niedobór androgenów lub estrogenów), tarczycy (obniżenie poziomu T3 , T4 , jodu związanego z białkami - PBI, gromadzenie 131 I przez tarczycę) i nadnerczy (obniżenie ilości kortyzolu i 17-OCS w osoczu, wydalanie 17-KC i 17-OCS w moczu, limfocytoza).
Wszystkie typy przysadkowej (podwzgórzowo-przysadkowej) karłowatości genetycznej charakteryzują się powtarzającą się chorobą dzieci w rodzinie z dziedziczeniem autosomalnym recesywnym (częściej) lub autosomalnym dominującym, zahamowaniem wzrostu i rozwoju fizycznego od 2-4 lat z opóźnieniem co najmniej 2-3 o od średnich norm wzrostu dla danej płci, wieku, populacji, z niską spontaniczną roczną dynamiką wzrostu, opóźnionym kostnieniem. Przy niskim poziomie hormonu somatotropowego (w 2-3 testach pobudzających poniżej 7 ng/ml) terapia hormonem somatotropowym jest wysoce skuteczna (daje wzrost wzrostu co najmniej 7 cm rocznie). Przy normalnym lub wysokim poziomie hormonu somatotropowego (przy jego biologicznej nieaktywności) wrażliwość na hormon może być zachowana. Nie obserwuje się zmian w inteligencji
W karłowatości genetycznej z tkankową niewrażliwością na hormon somatotropowy obraz kliniczny jest podobny do izolowanego niedoboru hormonu wzrostu, ale terapia somatotropiną jest nieskuteczna. W tej grupie można wyróżnić następujące główne formy według poziomu IRF: z prawidłową zawartością (defekt receptora IRF) i obniżoną - karłowatość typu Larona (niedobór IRF-1 i IRF-II) oraz typ występujący u afrykańskich pigmejów (niedobór IRF-1).
Karłowatość mózgowa charakteryzuje się występowaniem izolowanych chorób w rodzinie, związanych z wewnątrzmacicznym lub poporodowym uszkodzeniem ośrodkowego układu nerwowego, obecnością oczywistych zmian organicznych w ośrodkowym układzie nerwowym, często w połączeniu z patologią narządu wzroku, obecnością moczówki prostej, zachowaniem funkcji gonadotropowych i zmianami inteligencji.
Niektóre typy dysgenezji gonad i agenezji są związane ze znacznym niskim wzrostem, w szczególności zespół Shereshevsky-Turnera i „Turneroidowa” (mozaikowa) forma zespołu dysgenezji jąder. Badania cytogenetyczne (chromatyna płciowa, kariotyp) pomagają w diagnostyce różnicowej, ujawniając defekty chromosomowe, a także charakterystyczne defekty rozwoju somatycznego i płciowego, prawidłowe lub podwyższone poziomy endogennego hormonu somatotropowego i niewrażliwość na leczenie somatotropiną.
Spośród zaburzeń endokrynologicznych występujących przy niskim wzroście należy wyróżnić niedoczynność pierwotną tarczycy, spowodowaną wrodzoną hipoplazją lub aplazją tarczycy, jej dystopią, defektami enzymatycznymi w biosyntezie hormonów tarczycy, wczesnym autoimmunologicznym uszkodzeniem tarczycy. We wszystkich tych stanach dominują objawy niedoczynności tarczycy z wysokim poziomem TSH, spadkiem T4 i T3 w surowicy krwi . W obrzęku śluzowatym o podłożu autoimmunologicznym we krwi wykrywa się przeciwciała przeciwko tyreoglobulinie, frakcjom mikrosomalnym i jądrowym tkanki tarczycy, poziom hormonu somatotropowego jest prawidłowy lub obniżony. Efekt kliniczny można uzyskać, kompensując jedynie niedoczynność tarczycy.
Niski wzrost towarzyszy przedwczesnemu rozwojowi płciowemu i zespołowi nadnerczowo-płciowemu z powodu przedwczesnego zamknięcia stref wzrostu; choroba Itsenko-Cushinga, która występuje w dzieciństwie z powodu hamującego działania glikokortykoidów na wydzielanie hormonu somatotropowego i ich katabolicznego działania; zespół Mauriaca - niski wzrost i infantylizm pacjentów z ciężką cukrzycą insulinozależną.
Karłowatość przysadkową należy różnicować z opóźnieniem somatogennym rozwoju fizycznego spowodowanym przewlekłymi zaburzeniami metabolicznymi (w chorobach wątroby, nerek, przewodu pokarmowego), przewlekłym niedotlenieniem (w chorobach układu sercowo-naczyniowego, oddechowego, niedokrwistości); z chorobami układowymi układu ruchu (chondrodystrofia, niedoskonała osteogeneza, choroba zwyrodnieniowa stawów) itp.
Funkcjonalne (konstytucyjne) zahamowanie wzrostu jest czasami obserwowane wraz z późnym początkiem dojrzewania u pozornie zdrowych nastolatków; odkryliśmy, że jest ono przede wszystkim związane z przejściową niewydolnością aktywności gonadotropowej. Wydzielanie hormonu somatotropowego zwykle nie jest upośledzone lub jest nieznacznie zmniejszone. Stymulacja gonadotropin może przyspieszyć zarówno rozwój płciowy, jak i wzrost.
Niski wzrost o charakterze rodzinnym należy traktować jako odmianę rozwoju fizjologicznego.
Co trzeba zbadać?
Jak zbadać?
Jakie testy są potrzebne?
Z kim się skontaktować?
Leczenie karłowatość
Leczenie karłowatości to długi proces. Zmusza to lekarza do rozłożenia środków wpływających na wzrost w czasie, aby uzyskać jak największy efekt kliniczny, przestrzegając jednocześnie 2 podstawowych zasad:
- maksymalne zbliżenie rozwoju wywołanego leczeniem do warunków fizjologicznych;
- oszczędzając strefy wzrostu nasad kości.
Wieloletnie doświadczenie w leczeniu karłowatości pozwala nam uznać następujący schemat terapii etapowej za właściwy. Rozpoznanie karłowatości u pacjentów dorosłych zazwyczaj nie budzi wątpliwości. U małych dzieci, jeśli obraz kliniczny jest niejasny, konieczny jest okres diagnostyczny: 6-12 miesięcy obserwacji bez terapii hormonalnej. W tym czasie zaleca się kompleksowe leczenie wzmacniające ogólne; odpowiednie odżywianie ze zwiększeniem zawartości białka zwierzęcego, warzyw i owoców w diecie, witamin A i D, preparatów wapniowo-fosforanowych. Brak wystarczających zmian w rozwoju fizycznym i wzrostowym na tym tle oraz wykrycie zaburzeń endokrynologicznych podczas badania stanowią podstawę do rozpoczęcia terapii hormonalnej.
Głównym rodzajem terapii patogenetycznej karłowatości przysadkowej jest stosowanie ludzkiego hormonu wzrostu, ponieważ występowanie większości przypadków karłowatości jest niewątpliwie zależne od jednej lub drugiej formy jego niedoboru. Ze względu na specyfikę gatunkową tego hormonu, dla ludzi aktywna jest tylko somatotropina ludzka i naczelnych. W klinice szeroko stosowany jest lek wyizolowany z przysadki mózgowej osób zmarłych na choroby niezakaźne i nienowotworowe. Ludzką somatotropinę uzyskuje się poprzez syntezę bakteryjną z wykorzystaniem Escherichia coli metodą inżynierii genetycznej. Ludzką somatotropinę syntetyzuje się również chemicznie, ale jest ona niezwykle droga i praktycznie nie jest stosowana w klinice. Do leczenia somatotropiną wybiera się pacjentów ze stwierdzonym niedoborem endogennego hormonu wzrostu, z różnicowaniem szkieletowym nieprzekraczającym poziomu typowego dla wieku 13-14 lat. Nie ma ograniczeń wiekowych w leczeniu.
Minimalne skuteczne dawki, które można stosować w pierwszym okresie leczenia, to 0,03-0,06 mg/kg masy ciała. Najskuteczniejsze dawki to 2-4 mg 3 razy w tygodniu. Zwiększenie dawki pojedynczej do 10 mg nie wiązało się z odpowiednim zwiększeniem efektu wzrostu, ale powodowało szybkie tworzenie się przeciwciał przeciwko somatotropinie.
W naszym kraju prace nad badaniem ludzkiego hormonu wzrostu prowadzone są od 1960 roku. Testowano dwa schematy leczenia: ciągły i przerywany z kursami 2-3 miesięcznymi i takimi samymi przerwami między nimi. Średni przyrost wzrostu pacjentów w pierwszym roku leczenia wyniósł 9,52 ± 0,39 cm, przyrost masy ciała 4,4 ± 0,14 kg. Przy długotrwałym ciągłym leczeniu średni przyrost wzrostu wyniósł 0,82 cm/miesiąc, masa ciała - 0,38 kg/miesiąc; przy przerywanym - odpowiednio 0,75 cm/miesiąc i 0,4 kg/miesiąc. Ciągłe leczenie zapewniało szybszy przyrost wzrostu z gwałtownym spadkiem efektu po 1-1,5 roku, przy przerywanym leczeniu skuteczność utrzymywała się przez 3-4 lata, co pozwala nam uznać schemat leczenia kursowego za bardziej odpowiedni. Oznaczenie poziomu IGF-I (somatomedyny C) może służyć jako wiarygodny wskaźnik wrażliwości pacjenta na leczenie lekami somatotropowymi. Wzrost zawartości IGF-I po wprowadzeniu hormonu somatotropowego pozwala przewidzieć pozytywny efekt terapii. Ważną zaletą leczenia somatotropiną jest brak przyspieszenia kostnienia szkieletu na jego tle.
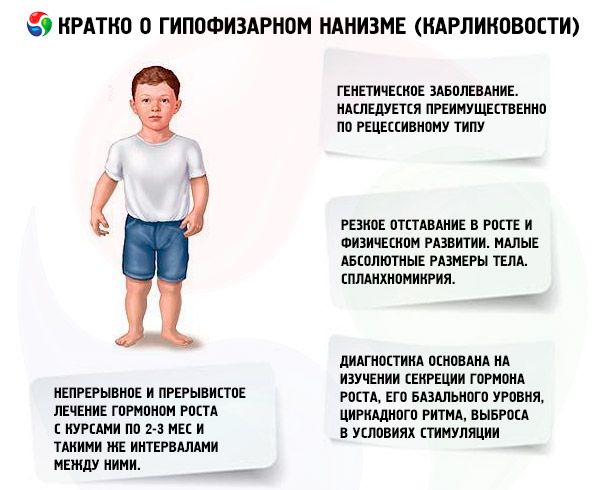
Najważniejszym sposobem leczenia karłowatości jest stosowanie sterydów anabolicznych, które stymulują wzrost poprzez zwiększenie syntezy białek i zwiększenie poziomu endogennego hormonu somatotropowego. Leczenie prowadzi się przez kilka lat, stopniowo zastępując niektóre leki innymi, ze związków mniej aktywnych na bardziej aktywne. Zmiana leków anabolicznych jest wskazana, gdy efekt wzrostu słabnie po 2-3 latach, co prowadzi do dodatkowego zwiększenia wzrostu. Leczenie prowadzi się w kursach (okres odpoczynku powinien wynosić połowę okresu leczenia). W przypadku uzależnienia wskazane są również dłuższe przerwy (do 4-6 miesięcy). Jednorazowo przepisuje się tylko jeden ze sterydów anabolicznych. Łączenie 2 lub więcej leków jest nieodpowiednie, ponieważ nie wzmacnia to ich efektów metabolicznych i wzrostowych. To ostatnie zależy przede wszystkim od wieku pacjentów i stopnia zróżnicowania kości szkieletowych na początku leczenia. Najlepszy efekt obserwuje się u pacjentów poniżej 16-18 roku życia z kostnieniem szkieletowym nieprzekraczającym poziomu charakterystycznego dla 14-latka. Wskazane jest rozpoczęcie leczenia natychmiast po postawieniu diagnozy, zwykle w wieku 5-7 lat. Przed leczeniem należy unikać przepisywania gonadotropin i hormonów płciowych, które stymulując wzrost, jednocześnie przyspieszają różnicowanie szkieletowe. Zasada dawkowania sterydów anabolicznych jest od minimalnych skutecznych dawek do stopniowo zwiększanych. Zalecane dawki najczęściej stosowanych leków: nerobol (metandrostenol, dianabol) - 0,1-0,15 mg na 1 kg masy ciała na dobę doustnie; nerobolil (durabolin) - 1 mg na 1 kg masy ciała na miesiąc domięśniowo, dawkę miesięczną podaje się w 2-3 dawkach odpowiednio po 15 lub 10 dniach; retabolil (deca-durabolin) - 1 mg na 1 kg masy ciała raz w miesiącu domięśniowo. Przekroczenie określonych dawek może prowadzić do androgenizacji. W dawkach fizjologicznych związki te nie wpływają znacząco na stan narządów płciowych i różnicowanie kości szkieletowych, co pozwala na ich długotrwałe stosowanie u pacjentów obu płci. Dziewczęta powinny znajdować się pod nadzorem lekarza ginekologa, ponieważ w przypadku przedawkowania lub zwiększonej indywidualnej wrażliwości u niektórych pacjentów mogą wystąpić objawy wirylizacji, które szybko ustępują po przerwaniu leczenia. Leki doustne metylowane do etylowanych w 17. pozycji mogą czasami powodować działanie cholestatyczne, dlatego w chorobach wątroby należy preferować pozajelitowe związki anaboliczne lub leki doustne należy łączyć z lekami żółciopędnymi. Bardzo rzadko leczenie sterydami anabolicznymi może powodować reakcje alergiczne (swędzenie, wysypki). W przypadku braku powikłań sterydy anaboliczne stosuje się tak długo, jak długo utrzymuje się efekt wzrostu (do 16-18 lat, a czasami dłużej). Leczenie prowadzi się na tle ogólnej terapii wzmacniającej.
Jeżeli pacjent ma objawy niedoczynności tarczycy, przepisuje się jednocześnie leki tarczycowe (tyroksynę, tyreoidynę, tyreotom) w indywidualnie dobranych dawkach.
W leczeniu chłopców kolejnym krokiem jest podawanie gonadotropiny kosmówkowej. Lek ten stosuje się nie wcześniej niż w wieku 15-16 lat, a często nawet później, aby stymulować komórki Leydiga, co przyspiesza zarówno rozwój płciowy, jak i wzrost (dzięki anabolicznej aktywności własnych androgenów). Dawki od 1000 do 1500 IU stosuje się 1-2 razy w tygodniu domięśniowo w 2-miesięcznych kursach nie częściej niż 2-3 razy w roku. Jeśli efekt jest niepełny, leczenie gonadotropiną kosmówkową u chłopców w wieku 16 lat i starszych jest przeplatane podawaniem małych dawek androgenów (metylotestosteronu w dawce 5-10 mg/dobę podjęzykowo).
Dziewczęta powyżej 16 roku życia mogą rozpocząć leczenie małymi dawkami estrogenów, symulując normalny cykl seksualny. Leczenie prowadzi się przez 3 tygodnie każdego miesiąca, po czym następuje przerwa. W 2. fazie cyklu, od 3. tygodnia, można przepisać gonadotropinę kosmówkową w dawce 1000-1500 IU 3-5 razy w tygodniu lub leki o działaniu gestagennym (pregnina, progesteron).
Ostatnim etapem leczenia (po zamknięciu stref wzrostu) jest stałe podawanie terapeutycznych dawek hormonów płciowych odpowiadających płci pacjenta, w celu pełnego rozwoju narządów płciowych, drugorzędnych cech płciowych, zapewnienia libido i potencji seksualnej. Preparaty skojarzone estrogenowo-progestagenowe (non-ovlon, bisecurin, infekundin, rigevidon) są wygodne w leczeniu pacjentek, a preparaty androgenowe o przedłużonym uwalnianiu (testenate, sustanon-250, omnadren-250) są wygodne w leczeniu pacjentów płci męskiej.
Przeprowadza się ogólne leczenie wzmacniające (reżim, dieta białkowo-warzywna, witaminoterapia, biostymulatory). Wskazane jest stosowanie preparatów cynkowych, w mechanizmie działania których główną rolę odgrywa zwiększenie aktywności IGF-1 (insulinopodobnego czynnika wzrostu I).
W przypadku patologii organicznej ośrodkowego układu nerwowego stosuje się terapię przeciwzapalną, resorpcyjną i odwadniającą. Celowana terapia systemowa daje zachęcający efekt. W wyniku długotrwałego leczenia etapowego 148 (80,4%) z 175 pacjentów z karłowatością obojga płci udało się osiągnąć wzrost ponad 130 cm, 92 (52,5%) - ponad 140 cm, a 32 (18,3%) - 150-160 cm i więcej. Jednocześnie 37 pacjentów (21,2%) zwiększyło swój wzrost o 30 cm, 107 (61,1%) o 31-50 cm, a 31 (17,7%) o 51-60 cm i więcej.
Prognoza
Rokowanie zależy od formy karłowatości. W genetycznych typach karłowatości rokowanie na całe życie jest korzystne. W przypadku obecności guza przysadki i organicznego uszkodzenia ośrodkowego układu nerwowego jest ono determinowane dynamiką rozwoju głównego procesu patologicznego. Nowoczesne metody terapii znacznie zwiększyły możliwości fizyczne i wydolność operacyjną pacjentów, a także wydłużyły ich oczekiwaną długość życia. W okresie aktywnego leczenia pacjenci muszą być badani przez lekarza co 2-3 miesiące, przy terapii podtrzymującej - co 6-12 miesięcy.
Zatrudnienie pacjentów odpowiadające ich możliwościom intelektualnym i fizycznym ma ogromne znaczenie dla ich adaptacji społecznej.
Wskazane jest wybieranie zawodów, które nie wiążą się z dużym wysiłkiem fizycznym, ale pozwalają wykazać się sprawnością intelektualną, umiejętnością wykonywania prac precyzyjnych i znajomością języków obcych.