
Priony - czynniki wywołujące choroby prionowe
Ostatnia recenzja: 23.04.2024

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Powolne infekcje wirusowe charakteryzują się specjalnymi kryteriami:
- niezwykle długi czas inkubacji (miesiące, lata);
- rodzaj porażki narządów i tkanek, głównie ośrodkowego układu nerwowego;
- powolny stały postęp choroby;
- nieunikniony wynik śmiertelny.

Niektóre patogeny ostrych infekcji wirusowych mogą również powodować powolne infekcje wirusowe. Na przykład, wirus odry czasami powoduje PSES, a wirus różyczki - postępującą wrodzoną różyczkę i różyczkę.
Typową powolną infekcją wirusową zwierząt jest wirus visna / medi, który należy do retrowirusów. Jest czynnikiem sprawczym powolnej infekcji wirusowej i postępującego zapalenia płuc u owiec. Biała istota mózgu zostaje zniszczona , rozwija się paraliż (visna - więdnięcie); występuje przewlekłe zapalenie płuc i śledziony.
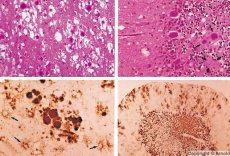
Podobne choroby spowodowane powolnymi infekcjami wirusowymi powodują priony - patogeny zakażeń prionami. Choroby prionowe to grupa postępujących zaburzeń OUN ludzi i zwierząt. Ludzie zaburzyli funkcjonowanie ośrodkowego układu nerwowego, nastąpiły zmiany osobowości, zaburzenia ruchowe. Objawy choroby zwykle trwają od kilku miesięcy do kilku lat, kończąc się śmiercią. Wcześniej, infekcje prionem były rozważane razem z tak zwanymi patogenami powolnych infekcji wirusowych.
Niektóre czynniki powodujące choroby prionowe gromadzą się najpierw w tkankach limfoidalnych. Priony docierające do mózgu, akumuluje się w dużych ilościach, co powoduje amyloidoza (zewnątrzkomórkowej disproteinoz charakteryzuje się odkładaniem amyloidu z rozwojem atrofii i stwardnienie tkanki) i astrocytozy (astrocytów proliferacją komórek glejowych, giperprodukdiya glialnyh włókien). Włókna lub agregaty amyloidu i zmiany gąbczaste mózgu (przenoszone encefalopatia). W rezultacie zmiany zachowania, koordynacja ruchów zostaje zakłócona, rozwija się wyczerpanie z fatalnym skutkiem. Odporność nie powstaje. Choroby związane z prionami chorób konformacyjnych, które rozwijają się w wyniku nieprawidłowego fałdowania (naruszenie właściwej konformacji) białka komórkowego wymagane dla normalnego funkcjonowania organizmu. Sposoby przenoszenia prionów są różnorodne:
- droga pokarmowa - zainfekowane produkty pochodzenia zwierzęcego, suplementy diety z surowych narządów bydlęcych itp.:
- transmisja z transfuzją krwi, podawanie produktów pochodzenia zwierzęcego, przeszczepianie narządów i tkanek, stosowanie zainfekowanych narzędzi chirurgicznych i dentystycznych;
- przekazywanie za pośrednictwem leków immunobiologicznych (wiadomo, że zakażają prpP '' '1500 owiec formulvaccine od chorych owiec).
Patologiczne priony, po dostaniu się do jelit, są transportowane do krwi i limfy. Po replikacji obwodowej w śledzionie, wyrostku robaczkowym, migdałkach i innych tkankach limfoidalnych są przenoszone do mózgu za pośrednictwem nerwów obwodowych (neuroinwazji). Prawdopodobnie bezpośrednia penetracja prionów do mózgu przez barierę krew-mózg. Wcześniej sądzono, że centralny układ nerwowy jest jedyną tkanką, w której gromadzą się patologiczne priony, ale pojawiły się badania, które zmieniły tę hipotezę. Okazało się, że gromadzenie prionów w śledzionie wiąże się ze wzrostem i funkcjonowaniem pęcherzykowych komórek dendrytycznych.
 [
[Właściwości prionów
Komórkową normalną izoformę białka prionowego o masie cząsteczkowej 33-35 kD określa gen białka prionu (gen prion-PrNP znajduje się na 20 chromosomie człowieka). Normalny gen pojawia się na powierzchni komórki (zakotwiczony w błonie przez cząsteczkę glikoproteiny), jest wrażliwy na proteazę. Reguluje przekazywanie impulsów nerwowych, cykl dobowy, procesy utleniania, bierze udział w metabolizmie miedzi w ośrodkowym układzie nerwowym oraz w regulacji podziału komórek macierzystych szpiku kostnego. Ponadto, gen prionu znajduje się w śledzionie, węzłach chłonnych, skórze, GIT i komórkach dendrytycznych pęcherzyków.
Proliferacja patologicznych prionów
Transformacja prionów w zmienione formy ma miejsce, gdy zaburzona jest kinetycznie kontrolowana równowaga między nimi. Proces zwiększa się, gdy wzrasta ilość patologicznego (PrF) lub egzogennego priona. PrP jest normalnym białkiem zakotwiczonym w błonie komórkowej. PrP 'jest kulistym białkiem hydrofobowym, które tworzy agregaty z samym sobą i z PrF "na powierzchni komórki: w rezultacie PrP" przekształca się w PrF "i cykl trwa. Patologiczna forma PrF "" gromadzi się w neuronach, nadając komórce gąbczasty wygląd.
Kuru
Choroba prionowa, wcześniej rozpowszechniona wśród papuasów (tłumaczona jako drżenie lub trzęsienie) we wschodniej części wyspy Nowej Gwinei. Zakaźne właściwości choroby udowodnił K. Gaidushek. Czynnik sprawczy jest przenoszony przez żywność w wyniku rytualnego kanibalizmu - jedzenia niedostatecznie przetworzonych termicznie, zainfekowanych prionów martwych krewnych mózgu. W wyniku klęski centralnego układu nerwowego zakłócają się ruchy, chód, dreszcze, euforia ("śmiejąca się śmierć"). Okres inkubacji trwa 5-30 lat. Rok później pacjent umiera.
Choroba Creutzfeldta-Jakoba
Choroby prionowe, która przepływa w postaci otępienia, wizualne i zaburzeń mózgowych i zaburzeń ruchowych śmiertelnej choroby w ciągu 4-5 miesięcy, w klasycznym wariantu choroby Creutzfeldta-Jakoba i (3-14 miesięcy, gdy nowy wariant choroby Creutzfeldta-Jakoba. Okres inkubacji może wynosić 20. Istnieją różne sposoby infekcji i przyczyny choroby:
- w przypadku stosowania niewystarczająco przetworzonych termicznie produktów pochodzenia zwierzęcego, na przykład mięsa, krów mózgowych, pacjentów z bydlęcą encefalopatią gąbczastą;
- Przeszczep tkanki, np rogówki, transfuzję krwi, stosowanie hormonów i innych substancji biologicznie czynnych pochodzenia zwierzęcego, stosowanie katgut lub niedostatecznie prosterilinovannyh zanieczyszczonych instrumentów chirurgicznych, manipulacji wypreparowującym;
- do hiperprodukcji PrP i innych stanów, które stymulują proces transformacji PrP 'do PrF. "
Choroba może również rozwijać się w wyniku mutacji lub insercji w regionie genu prionu. Rodzinny charakter choroby jest powszechny w wyniku genetycznej predyspozycji do choroby Creutzfeldta-Jakoba. W przypadku nowego wariantu choroby Creutzfeldta-Jakoba zaburzenia rozwijają się w młodszym wieku (średni wiek 28 lat), w przeciwieństwie do wariantu klasycznego (średni wiek 65 lat). W nowym wariancie choroby Creutzfeldta-Jakoba nieprawidłowe białko prionowe gromadzi się nie tylko w ośrodkowym układzie nerwowym, ale także w tkankach limfo-rurgicznych, w tym w migdałkach.
Syndrom Gerstmanna-Streusslera-Sheinkera
Dziedziczna choroba prionowa, która występuje przy otępieniu, niedociśnieniu, połykaniu (dysfagia), dyzartrii. Często nosi charakter rodzinny. Okres inkubacji wynosi od 5 do 30 lat. Choroba występuje po 50-60 latach, jej czas trwania wynosi od 5 do 13 lat.
Dziedziczna śmiertelna bezsenność
Autoimmunologiczną chorobą postępującą bezsenność, współczulnego oskrzeli (nadciśnienie, gorączka, wysypka, tachykardia), drżenie, ataksja, mnokloniyami, halucynacje. Sen jest mocno zakłócony. Śmierć pojawia się wraz z postępem niewydolności sercowo-naczyniowej.
Napraw to
Scrapie (z angielskiego - złom. Ścierania) - choroby prionowe owiec i kóz (skórnego), płynącego z chorobami OUN, postępujące zaburzenia ruchu, silne swędzenie (świądu) i kończące się śmiercią zwierząt.
Gąbczasta encefalopatia bydła
Choroba bydła, charakteryzująca się porażką ośrodkowego układu nerwowego, naruszeniem koordynacji ruchów i nieuchronną śmiercią zwierzęcia. Po raz pierwszy epidemia choroby wybuchła w Wielkiej Brytanii. Było to związane z karmieniem zwierząt mączką mięsno-kostną zawierającą patologiczne priony. Okres inkubacji waha się od 1,5 do 15 lat. Najbardziej zainfekowanymi są głowa, rdzeń kręgowy i gałki oczne zwierząt.
Diagnostyka laboratoryjna chorób prionowych
Kiedy zdiagnozowane, gąbczaste zmiany w mózgu, astrocytoza (glejoza), brak nacieków zapalnych. Mózg jest zabarwiony amyloidem. W płynie mózgowo-rdzeniowym wykrywa się markery białkowe zaburzeń mózgu prionów (za pomocą testu ELISA). Przeprowadzono analizę genetyczną genu prionowego (PCR).
Profilaktyka chorób prionowych
Odkażania narzędzi i przedmiotów środowisku zalecane w autoklawie (w temperaturze 134 ° C, 18 min w temperaturze 121 ° C przez 1 godzinę)., Pieczenie, dodatkowe przetwarzanie wybielacza i roztworu odnonormalnym NaCl w ciągu 1 godziny dla niespecyficznej zapobiegania nakłada ograniczenia na stosowanie produktów leczniczych, pochodzenia zwierzęcego, a zabrania produkcję hormonów przysadki mózgowej zwierząt. Ograniczyć przeszczep opony twardej. Podczas pracy z płynami pacjenci dialogicznego używać rękawic gumowych.