Ekspert medyczny artykułu

Nowe publikacje
hamartoma
Ostatnia recenzja: 29.06.2025

Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
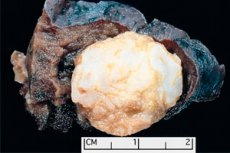
Guzowaty twór zlokalizowany w dowolnym obszarze anatomicznym, będący wynikiem nieprawidłowego rozrostu łagodnej tkanki, w medycynie określany jest mianem hamartoma (od greckiego hamartia – błąd, wada). [ 1 ]
Epidemiologia
Statystycznie, hamartomas stanowią 1,2% łagodnych nowotworów. Częstość występowania hamartomas płuc szacuje się na około 0,25% populacji ogólnej i stanowi do 8% wszystkich nowotworów płuc. Większość hamartomas płuc jest diagnozowana przypadkowo u pacjentów w wieku od 40 do 70 lat, ale są bardzo rzadkie w praktyce pediatrycznej.
Ogólnie rzecz biorąc, guzy hamartomatyczne diagnozuje się u mężczyzn, aczkolwiek w przypadku nerek występują częściej u kobiet i są diagnozowane w średnim wieku.
Około 5% łagodnych guzów piersi stanowią guzy hamartomatyczne. Najczęściej występują u kobiet po 35. roku życia.
80-90% guzów hamartomatycznych mózgu i ponad 50% guzów hamartomatycznych serca wiąże się ze stwardnieniem guzowatym.
Przyczyny hamartomy
Gamartomy należą do wad wrodzonych i są formacjami o łagodnym charakterze, które powstają z tkanek mezenchymalnych pochodzących z listków zarodkowych. A przyczyny ich występowania są związane z niekontrolowanym podziałem komórek cytologicznie prawidłowych tkanek (łącznej, mięśni gładkich, tłuszczu lub chrząstki), charakterystycznych dla danej lokalizacji anatomicznej, oraz ich ogniskowym przerostem podczas embriogenezy niemal każdego narządu lub struktury anatomicznej.
Występowanie wielu guzów hamartomatycznych u tego samego pacjenta określa się często mianem hamartomatozy lub hamartoma plejotropowego.
Guzy te mogą występować sporadycznie lub w obecności pewnych chorób dziedziczonych w sposób autosomalny dominujący, a także zespołów chorobowych uwarunkowanych genetycznie.
W wielu przypadkach guzy hamartomatyczne powstają, gdy rzadka choroba genetyczna o charakterze wielonarządowym – stwardnienie guzowate – ujawnia się wkrótce po urodzeniu lub w rodzinnej chorobie Recklinghausena – nerwiakowłókniakowatości typu 1. [ 2 ]
Czynniki ryzyka
Do głównych czynników ryzyka powstawania guzów hamartomatycznych zalicza się występowanie w historii choroby tzw. zespołów genetycznych polipowatości hamartomatycznej, w tym:
- Zespół mnogich guzów hamartomatycznych - zespół Cowden, w którym tworzą się liczne guzy hamartomatyczne pochodzenia ekto-, ento- i mezodermalnego, obserwuje się polipowatość przewodu pokarmowego oraz objawy śluzówkowo-skórne;
- Zespół Peutza-Jeghersa-Turena (charakteryzuje się rozwojem łagodnych polipów hamartomatycznych w przewodzie pokarmowym);
- Zespół Proteusza;
- Zespół Weila - młodzieńcza polipowatość jelita grubego;
- Zespół Bannayana-Rileya-Ruvalcaby, który podobnie jak zespół Cowdena powoduje powstawanie licznych hamartoma (polipów hamartomatycznych) jelita;
- Zespół Carneya-Stratakisa i kompleks Carneya.
Ponadto guzy hamartomatyczne tworzą się u chorych z dziedzicznym zespołem Watsona oraz w przypadkach sporadycznie występującego lub wrodzonego zespołu Pallistera-Halla z hamartomą podwzgórza i polidaktylią.
Patogeneza
Mechanizm wzmożonej proliferacji tkanek zarodkowych, prowadzącej do powstawania zmian nowotworowych w różnych narządach, tłumaczony jest aberracjami chromosomowymi i mutacjami genów, które mogą występować spontanicznie lub być dziedziczne.
W stwardnieniu guzowatym zidentyfikowano mutacje w genach TSC1 lub TSC2 - supresorach nowotworów, które zapobiegają i hamują nadmierną proliferację - zbyt szybki lub niekontrolowany wzrost i podział komórek. A w neurofibromatozie typu 1 i zespole Watsona - mutacje linii zarodkowej genu supresora nowotworu mitochondrialnego NF1.
W zespole guza hamartomatycznego, który łączy w sobie zespoły Cowden, Protea, Bannayana-Rileya-Ruvalcaby i polipowatości młodzieńczej, patogeneza wiąże się z mutacją genu PTEN, który koduje enzym biorący udział w regulacji proliferacji i jest uważany za gen supresorowy guza.
Mutacje w genie STK11 kodującym strukturę i funkcję jednego z transbłonowych enzymów serynowych, co zmniejsza jego zdolność do hamowania podziału komórek, prowadzą do zespołu Peutza-Jeghersa-Turena, z rozwojem polipów jelitowych i pigmentowanych zmian skórnych. Mutacja w genie GLI3, czynniku transkrypcyjnym zaangażowanym w tworzenie tkanki wewnątrzmacicznej, została zidentyfikowana w zespole Pallistera-Halla.
W rezultacie niekontrolowany wzrost komórek spowodowany mutacjami genów prowadzi do powstania guzów hamartomatycznych.
Objawy hamartomy
W zależności od umiejscowienia guzów hamartomatycznych rozróżnia się ich rodzaje, z których każdy ma swoją własną strukturę i symptomatologię.
Hamartoma płuca
Hamartoma płucna może powstać w dowolnym płacie i obwodowych częściach płuc i składa się z normalnych tkanek obecnych w płucach: tłuszczowej, nabłonkowej, włóknistej i chrzęstnej. W 80% przypadków przeważa składnik chrzęstny (komórki chrząstki szklistej) z włączeniem adipocytów - komórek tkanki tłuszczowej i komórek nabłonka dróg oddechowych. [ 3 ]
Wcześniejsze nazwy: chrzęstniak hamartoma, mezenchymoma, chrzęstniak hamartoma lub hamartochondroma nie są obecnie zalecane przez WHO.
Z kolei guzy mezenchymalne torbielowate hamartoma płuc występują rzadziej i u większości pacjentów wiążą się z zespołem Cowden.
Uszkodzenie hamartomatyczne płuc może nie dawać żadnych objawów, ale może powodować objawy w postaci przewlekłego kaszlu (często z krwiopluciem), świszczącego oddechu przy oddychaniu i trudności w oddychaniu. [ 4 ]
Hamartoma serca
Do łagodnych pierwotnych nowotworów serca u dorosłych zalicza się dojrzałe hamartoma mięśnia sercowego, a u niemowląt i dzieci ze stwardnieniem guzowatym – mięśniaka prążkowanokomórkowego, czyli hamartoma mięśnia sercowego komór lub przegrody międzykomorowej. [ 5 ]
Dojrzały kardiomiocyt hamartoma rozwija się w ścianie komory (i rzadko w przedsionkach) i może pojawić się jako wiele zmian, które są gęstymi masami ściśle związanymi z leżącym pod nimi mięśniem sercowym. Guz może powodować objawy niewydolności serca: ból w klatce piersiowej, kołatanie serca i arytmie, szmery serca, obrzęk, duszność, sinicę.
Mięśniaki prążkowanokomórkowe serca, z których większość diagnozuje się w pierwszym roku życia, składają się z tkanki mięśnia sercowego wytwarzanej przez mioblasty zarodkowe i mają wygląd litych mas ogniskowych bez otoczki.
Zazwyczaj guzy hamartomatyczne nie dają żadnych objawów i samoistnie zanikają przed ukończeniem 4 roku życia.
Niektórzy eksperci uważają również, że zmiany hamartomatyczne są związane z mięśniakiem serca typu Carneya. [ 6 ]
Gamartoma przewodu pokarmowego
Hamartoma żołądka to masa mezenchymalna w postaci nabłonkowego polipa hiperplastycznego żołądka, polipa Peutza-Jeghersa i rzadkiego hamartoma mioepithelialnego - z przerośniętymi pęczkami mięśni gładkich. Inne nazwy tego hamartoma to hamartoma mięśniowo-gruczołowa, hamartoma gruczolakowata i adenomyoma żołądka. Typowe objawy kliniczne obejmują niestrawność, ból w nadbrzuszu i krwawienie z górnego odcinka przewodu pokarmowego. [ 7 ], [ 8 ]
Więcej informacji w materiale - Polipowatość żołądka
Hamartoma jelitowa to polip hamartomatyczny lub hiperplastyczny jelita grubego, diagnozowany jako gruczolak gruczolakowaty lub cewkowy. Gdy hamartoma jest zlokalizowany w gruczole Brunnera dwunastnicy, objawy objawiają się bólem w okolicy nadbrzusza; nudnościami, wymiotami i wzdęciami (oznaczającymi niedrożność jelit); a jeśli jest znacznych rozmiarów, krwawieniem z przewodu pokarmowego. W przypadkach hamartoma mioepitelialnego jelita krętego pacjenci skarżą się na ból brzucha, mają zmniejszoną masę ciała i rozwijają przewlekłą anemię. [ 9 ], [ 10 ]
Przeczytaj także - Polipy odbytu
Hamartoma retrorektalna to torbielowata hamartoma lub wielokomorowa torbiel przestrzeni retrorektalnej (luźnej tkanki łącznej między odbytem a jego własną powięzią), która najczęściej występuje u kobiet w średnim wieku. Ma wygląd torbieli wybrzuszającej się z tylnej ściany odbytnicy, która jest wyłożona nabłonkiem i zawiera chaotycznie ułożone włókna mięśni gładkich. Ta hamartoma objawia się bólem dolnej części brzucha i nawracającymi zaparciami. [ 11 ], [ 12 ]
Guzy hamartomatyczne wątroby i śledziony
Wielokrotny hamartoma żółciowy wątroby to hamartoma międzywątrobowych dróg żółciowych związanych z wadami rozwojowymi w okresie embrionalnym. Ten hamartoma (pojedynczy lub wielokrotny) składa się z chaotycznie rozszerzonych skupisk dróg żółciowych i podścieliska włóknisto-kolagenowego. [ 13 ]
Guzy hamartomatyczne dróg żółciowych nie dają żadnych objawów i zwykle są wykrywane przypadkowo (podczas badania radiologicznego lub laparotomii). [ 14 ]
Rzadkim i często przypadkowo wykrywanym nowotworem pierwotnym o charakterze łagodnym jest hamartoma śledziony, który składa się z elementów czerwonej miazgi śledziony - w postaci dobrze zdefiniowanej jednorodnej masy o zwartej konsystencji. Malformacja ta może być pojedyncza lub mnoga; przy ściskaniu miąższu śledziony może wystąpić uczucie dyskomfortu i bólu w lewym podżebrzu. [ 15 ], [ 16 ]
Guzy hamartomatyczne nerek
Najczęstszym hamartomą nerki jest angiomyolipoma nerki, ponieważ ten łagodny guz składa się z dojrzałej tkanki tłuszczowej z osadzonymi włóknami mięśni gładkich i naczyniami krwionośnymi. Powstaje w stwardnieniu guzowatym w 40-80% przypadków. Zwiększenie rozmiaru hamartoma (ponad 4-5 cm) prowadzi do bólu i pojawienia się krwi w moczu. [ 17 ], [ 18 ]
Hamartoma piersi
Przyjęte przez WHO definicje diagnostyczne hamartoma piersi to takie terminy jak adenolipoma, chondrolipoma i hamartoma mięśniakowata. Chociaż mammolodzy często nazywają go fibroadenolipoma, ponieważ formacja guza zawiera komórki tkanki włóknistej, gruczołowej i tłuszczowej zamknięte w cienkiej torebce tkanki łącznej o wyraźnych zarysach. Podczas badania wzrokowego można zaobserwować ogniskowe zwapnienia. W tym przypadku objawy kliniczne są nieobecne. [ 19 ], [ 20 ]
Przeczytaj także - Guzy piersi
Guzy hamartomatyczne mózgu
U jednej trzeciej pacjentów ze stwardnieniem guzowatym występuje hamartoma mózgu w postaci wewnątrzczaszkowych wyrostków korowych lub guzków w różnych płatach - na granicy istoty szarej i białej - lub guzków podwyściółkowych wzdłuż ścian komór mózgowych. Może również powstać hamartoma astrocytarna, podwyściółkowy olbrzymiokomórkowy gwiaździak z zaburzeniem korowym, dysmorficznymi neuronami i dużymi komórkami glejowymi miąższu mózgu (astrocyty). Objawy hamartoma mózgu obejmują ataki padaczkowe i upośledzenie umysłowe u dzieci. [ 21 ], [ 22 ]
Rzadką wadą rozwojową, która występuje podczas embriogenezy i jest obecna od urodzenia, jest hamartoma podwzgórza, czyli masa heterotopowych neuronów i komórek glejowych. W miarę wzrostu mózgu dziecka guz powiększa się, ale nie rozprzestrzenia się na inne obszary mózgu. [ 23 ], [ 24 ]
Jeżeli w przedniej części podwzgórza (tuber cinereum), gdzie przylega do niego przysadka mózgowa, tworzą się przerośnięte tkanki, wówczas wada ta objawia się objawami przedwczesnego ośrodkowego rozwoju płciowego (przed 8-9 rokiem życia): pojawieniem się wysypki trądzikowej, przedwczesnym rozwojem gruczołów piersiowych i wczesną pierwszą miesiączką u dziewczynek; wczesnym owłosieniem łonowym i mutacją głosu u chłopców.
Gdy guzy hamartomatyczne tworzą się w tylnej części podwzgórza, mogą wystąpić nieprawidłowości w aktywności elektrycznej mózgu, które we wczesnym dzieciństwie objawiają się napadami padaczkowymi, a w późniejszym okresie (w wieku 4-7 lat) padaczką z ogniskowymi napadami padaczkowymi z nagłym śmiechem lub z mimowolnym płaczem, napadami atonicznymi i toniczno-klonicznymi, a także napadami agresji, problemami z pamięcią i funkcjami poznawczymi.
Guz przysadki jest sporadycznie występującym łagodnym gruczolakiem przysadki.
U dorosłych w średnim wieku z zespołem Cowdena może wystąpić rzadka masa przypominająca guz, hamartoma móżdżku, diagnozowana jako dysplastyczny gangliocytoma móżdżku lub choroba Lhermitte'a-Duclosa. Objawy mogą być nieobecne lub objawiać się bólem głowy, zawrotami głowy, upośledzoną koordynacją ruchów i porażeniem pojedynczych nerwów czaszkowych.
Węzeł chłonny hamartoma
W przypadku przerostu komórek mięśni gładkich i tkanki tłuszczowej, a także naczyń krwionośnych i podścieliska kolagenowego węzłów chłonnych pachwinowych, zaotrzewnowych, podżuchwowych i szyjnych powstaje guz hamartomatyczny naczyniakomięśniaka węzła chłonnego lub guzkowy hamartoma naczyniakomięśniaka – z częściową lub całkowitą wymianą jego miąższu. [ 25 ], [ 26 ]
Hamartoma skóry
W przypadku stwardnienia guzowatego lub nerwiakowłókniakowatości obserwuje się różne guzy hamartomatyczne skóry, najczęściej w postaci plam odbarwionych; plamy kawy i mleka; naczyniakowłókniaki (na policzkach, brodzie, bruzdach nosowo-wargowych); plamy szagrenowe o różnej lokalizacji (będące znamionami tkanki łącznej); włókniste blaszki na czole, skórze głowy lub szyi.
Rzadkim objawem dermatologicznym stwardnienia guzowatego (szczególnie u mężczyzn) jest guz mieszkowo-torbielowaty i kolagenowy hamartoma, charakteryzujący się obfitym odkładaniem się kolagenu w skórze właściwej, koncentrycznym włóknieniem okołomieszkowym i wypełnionymi keratyną podskórnymi torbielami o kształcie lejka, widocznymi w badaniu histopatologicznym. [ 27 ]
Do guzów hamartomatycznych składających się z melanocytów (komórek produkujących pigment melaniny) większość ekspertów zalicza także różne nowotwory melanocytowe, w szczególności wrodzone znamiona melanocytowe, które stanowią nieprawidłowość embriogenezy.
Pod względem etiologii hamartomy zbudowane z tkanki naczyniowej są także naczyniakami skóry.
U pacjentów z zespołem Peutza-Jeghersa-Thurena występuje hamartoma w postaci plamistej pigmentacji skóry i błon śluzowych - lentiginosis periorificialis
Przypadki liniowej grudkowej ektodermalnej i mezodermalnej hamartomy (Hamartoma moniliformis) charakteryzują się linijną, grudkową wysypką w kolorze skóry na głowie, szyi i górnej części klatki piersiowej.
A hamartoma łojotokowa to hamartoma gruczołów łojowych, o czym szerzej można przeczytać w publikacji - znamię łojowe.
Hamartoma oka
Pigmentowane zmiany hamartomatyczne tęczówki w neurofibromatozie typu 1 i zespole Watsona - w postaci guzkowych skupisk melanocytów dendrytycznych - są definiowane jako hamartoma tęczówki lub guzki Lischa. Są to przezroczyste (zwykle nie wpływające na widzenie) zaokrąglone, kopulaste, żółtobrązowe grudki, które wystają ponad powierzchnię tęczówki.
U chorych na młodzieńcze naczyniakowłókniaki nosogardła i rodzinną polipowatość gruczolakowatą często rozwija się mieszany hamartoma siatkówki i nabłonka barwnikowego siatkówki – w postaci czarnej plamy na centralnej (plamkowej) części siatkówki. [ 28 ]
Hamartoma nosa
Hamartoma nosa jest definiowana przez specjalistów jako chrzęstno-mięśniowy hamartoma nosa lub chrzęstniak nosa, spowodowany łagodną proliferacją nabłonka oddechowego, gruczołów podśluzówkowych i chrzęstno-kostnej tkanki łącznej. Jej objawy kliniczne zależą od wielkości i lokalizacji zmiany i obejmują: zatkany nos, trudności w oddychaniu przez nos i karmieniu piersią u niemowląt, przezroczystą, wodnistą wydzielinę z nosa i krwawienie z nosa. Hamartoma może rosnąć wraz z dzieckiem i rozprzestrzeniać się na oczodoły, powodując przesunięcie gałki ocznej do przodu lub do tyłu, zeza lub zaburzenia ruchowe oka. [ 29 ]
Hamartoma u dziecka
Wszystkie wyżej wymienione uszkodzenia hamartomatyczne różnych narządów i struktur anatomicznych występują u dzieci z odpowiadającymi im zespołami chorobowymi.
Noworodki mają hamartoma mezenchymalny ściany klatki piersiowej lub hamartoma chrzęstny żebra, które są litymi nieruchomymi masami powstałymi w wyniku ogniskowego przerostu normalnych elementów szkieletowych elementami chrzęstnymi, naczyniowymi i mezenchymalnymi. Ten hamartoma może powodować niewydolność oddechową i rozwój zespołu zaburzeń oddechowych. Hamartoma mezenchymalny wątroby jest drugim najczęściej występującym łagodnym guzem wątroby u dzieci. Ta formacja przypominająca guz (częściej zlokalizowana w prawym płacie narządu) składa się z komórek podścieliska mezenchymalnego, hepatocytów i komórek nabłonkowych wyściółki dróg żółciowych. Obraz kliniczny obejmuje wyczuwalną masę w jamie brzusznej, anoreksję i utratę masy ciała, a w przypadku znacznych rozmiarów (do 10 cm i więcej) guz obejmuje pozawątrobowe drogi żółciowe i żyłę główną dolną, co prowadzi do żółtaczki i obrzęków kończyn dolnych.
Hamartoma to wrodzony nerczak mezoblastyczny (występujący u 1 na 200 000 niemowląt), który może powodować wzdęcia brzucha u noworodka z wyczuwalną masą o gęstej konsystencji w prawym górnym kwadrancie brzucha. U niemowląt może również występować szybki, płytki oddech.
Do rzadkich wrodzonych anomalii należy włóknisty guz hamartomatyczny wieku niemowlęcego, który występuje u dzieci w pierwszych dwóch latach życia i objawia się bezbolesną, guzkowatą masą w tkance podskórnej pachy, szyi, barku i przedramienia, pleców i klatki piersiowej, uda, stopy i zewnętrznych narządów płciowych.
Eccrine angiomatous hamartoma u dziecka może być obecny od urodzenia lub ujawnić się we wczesnym dzieciństwie. Ten łagodny guz o charakterze hamartomatycznym zwykle ma wygląd niebieskawych lub brązowawych guzków i/lub blaszek, które są wynikiem proliferacji tkanki gruczołów potowych ekrynowych i naczyń włosowatych w środkowych i głębokich warstwach skóry właściwej. Ten hamartoma może powodować miejscową nadpotliwość i wzmożony wzrost włosów.
Komplikacje i konsekwencje
Ogólnie przyjmuje się, że hamartoma rzadko nawraca lub przekształca się w nowotwory złośliwe. Często wykazują niewielkie objawy lub nie wykazują ich wcale, a czasami nawet znikają z czasem. Jednak w cięższych przypadkach i w zależności od miejsca powstania, te wady rozwojowe mogą mieć poważne powikłania i konsekwencje.
Po pierwsze, hamartoma może osiągnąć takie rozmiary, że zaczyna uciskać otaczające tkanki i narządy, zaburzając ich funkcjonowanie.
Hamartoma serca u dzieci może prowadzić do trwałych zaburzeń rytmu serca, wad zastawek i upośledzenia przepływu krwi wewnątrzsercowej, a w konsekwencji do zastoinowej niewydolności serca.
Powikłaniami polipów hamartomatycznych przewodu pokarmowego są krwawienie z przewodu pokarmowego, niedrożność i intususcepcja jelit (z możliwym skutkiem śmiertelnym). A duży hamartoma nerki może wywołać pęknięcie nerki.
Hamartoma mózgu może być przyczyną zespołu wodogłowia obturacyjnego.
W przypadku guzów hamartomatycznych podwzgórza i przysadki mózgowej produkcja hormonu somatotropowego (hormonu wzrostu) może być upośledzona, co prowadzi do rozwoju niedoczynności przysadki mózgowej (hypotituitarism) u dzieci. U dzieci guzy hamartomatyczne podwzgórza mogą również prowadzić do lekoopornej padaczki.
Powikłania hamartoma nabłonka barwnikowego siatkówki wiążą się z dysfunkcją siatkówki i/lub nerwu wzrokowego, obrzękiem plamki, nowotworzeniem naczyń krwionośnych naczyniówki i odwarstwieniem siatkówki.
Diagnostyka hamartomy
Ważną częścią diagnostyki guzów hamartomatycznych i pokrewnych zespołów chorobowych jest zebranie wywiadu, w tym historii rodzinnej.
Badania laboratoryjne obejmują badania krwi: ogólne kliniczne; elektrolity w surowicy; profil limfocytów; poziomy wapnia, potasu, fosforanów i mocznika; oraz testy czynności wątroby. Jeśli to możliwe, wykonuje się biopsję aspiracyjną cienkoigłową masy, ponieważ badanie histologiczne ma kluczowe znaczenie w diagnozie i wyborze taktyki leczenia.
Diagnostyka instrumentalna pozwala na uwidocznienie guza hamartomatycznego i określenie jego dokładnej lokalizacji. Do tego celu wykorzystuje się badania rentgenowskie, angiografię, elektroencefalografię (EEG), ultrasonografię (USG), TK (tomografię komputerową), PET (pozytonową tomografię emisyjną), MRI (obrazowanie metodą rezonansu magnetycznego).
Diagnostyka różnicowa
W przypadku wszelkich nieprawidłowych mas bardzo ważna jest diagnostyka różnicowa. W ten sposób rozróżnia się gruźlika i hamartoma; hamartoma płuc i pierwotny rak płuc, rakowiaka oskrzeli, chorobę przerzutową. Hamartoma mózgu należy odróżnić od kraniopharyngioma i glejaka podwzgórzowo-skrzyżowaniowego. A diagnostyka różnicowa hamartoma jako wrodzonego nerczaka mezoblastycznego obejmuje guz Wilmsa (złośliwy nerczak zarodkowy), mięsaka jasnokomórkowego nerki i kostniejącego guza nerki u niemowląt.
Z kim się skontaktować?
Leczenie hamartomy
Jeśli hamartoma jest bezobjawowa i zostanie wykryta przypadkowo, nie jest wymagane żadne leczenie, ale konieczne jest monitorowanie jej „zachowania” i stanu pacjenta. W innych przypadkach terapia ma na celu zmniejszenie nasilenia objawów i zapobieganie powikłaniom. Na przykład w przypadku hamartoma podwzgórza z objawami przedwczesnego dojrzewania przepisuje się pewne leki, które hamują uwalnianie niektórych hormonów. Leki kardiologiczne są stosowane w celu leczenia objawów niewydolności serca u pacjentów z hamartoma serca.
Usunięcie guzów hamartomatycznych metodą chirurgiczną jest wskazane w celu potwierdzenia diagnozy oraz w przypadku niemożliwych do skorygowania medycznie, intensywnych objawów.
Na przykład, hamartoma płuc może zostać wycięta przez resekcję klinową, a w ciężkich przypadkach przez usunięcie płata płuca (lobektomia). Hamartoma piersi może zostać również wycięta, a jeśli jest duża, może być wymagana częściowa lub całkowita mastektomia.
Stereotaktyczna termoablacja radiofrekwencji lub ablacja laserowa mogą być stosowane do usuwania polipów hamartomatycznych. Radiochirurgia z silnie skupionymi promieniami gamma - nóż gamma do podwzgórzowych guzów hamartomatycznych lub astrocytarnych guzów hamartomatycznych - jest również stosowana.
Zapobieganie
Jedyną metodą zapobiegania rozwojowi guzów hamartomatycznych mogą być badania genetyczne przyszłych rodziców dziecka.
Prognoza
Ogólne rokowanie w przypadku tej wrodzonej anomalii zależy od umiejscowienia i rozmiaru nowotworu, a także od chorób współistniejących i ogólnego stanu zdrowia pacjenta.